In principle, siRNA could be designed to silence any pathological transcript to a high degree of base-pair precision. In practice, the scorecard is decidedly one-sided: a handful of hepatic triumphs and a very long list of extra-hepatic failures that never even got to the patient. The problem is not one of molecular ineptitude but anatomic exclusion. Naked siRNA is filtered, degraded, opsonized and repelled long before it reaches the cytosol of the intended cell. Highly optimized nanoparticle systems reverse this process by addressing the delivery challenge as a multi-barrier negotiation rather than a single membrane to be crossed. By tuning size, charge and surface-chemistry in an iterative manner, they avoid renal extraction, cloak themselves from reticulo-endothelial sentinels, exploit vascular heterogeneity to reach target organs, and then self-destruct in response to endosomal acidity to co-locate intact duplexes next to the RISC. The remaining obstacle is to repeat this dance in every patient, every dose, without inducing neutralizing antibodies or off-target cytokine storms.
The appeal of siRNA therapeutics is straightforward: given the address of any gain-of-function allele, viral transcript or oncogenic fusion, a 19-mer duplex can be designed in silico overnight to switch it off regardless of the chemistry of its binding pocket or enzymatic cleft. This simple yet powerful idea encounters a complex physiological landscape in which a molecule is too small to be retained in plasma yet too large to pass through endothelial fenestrae, too anionic to approach a membrane yet too immunogenic to be ignored. Early clinical efforts attempted to overcome this by brute force: dosing the circulation with milligram quantities of the molecule, only to find that the excess duplex would saturate the endogenous RNAi machinery, saturate renal clearance pathways and saturate patient tolerance to infusion reactions. The modern era of these therapies therefore accepts that the payload is innocent but the passport is flawed: success is no longer measured in terms of gene knock-down in a reporter cell line but by the engineering of a delivery vector that can traverse anatomy, immunology and pharmacokinetics in concert.
RNAi is not an enzymatic event but a repression cascade. It starts with the recognition of a double-stranded RNA duplex by the cytoplasmic sensor Dicer, an RNase-III that processes the duplex to 21- to 23-nucleotide (nt) size to suit the asymmetric cavity of Argonaute-2, the catalytic component of RISC. Strand selection is thermodynamically biased: the less stably paired 5′ end of one of the strands is retained as guide, while the other strand is cleaved and ejected as passenger. The guide is dragged through messenger RNA populations by diffusion-coupled scanning. A contiguous seven-nt seed match nucleates an interaction that, if completed by further complementarity, aligns the target nucleotide opposite the catalytic Asp-Asp-His triad of Argonaute. Cleavage between positions 10 and 11 (numbered from the 5′ end of guide) yields a nicked transcript that is bound by exonucleases and then degraded beyond repair. The cycle is catalytic: the same guide strand will iterate through dozens of messenger molecules until cell division or chemical modification. This catalytic property is the aetiology of both the allure and the risk of therapeutic siRNA: off-target silencing is amplified in proportion to on-target. Moreover, the pathway is shared with innate immunity: perfect blunt-ended duplexes longer than siRNA canonical size can activate RIG-I, while GU-rich stretches are bound by TLR-7 and TLR-8 in endosomes, and both launch interferon cascades that can mask the targeted gene silencing by a global antiviral response. These intersections are therefore not an academic afterthought but a design imperative that informs every chemical modification, every overhang geometry and every delivery vehicle.
Delivery fails not at a single barrier but at a series of gates that stand watch over every tissue compartment. In plasma, naked siRNA is degraded by extracellular ribonucleases that prefer double-stranded substrates, with an effective half-life of less than a circulatory pass. The kidney filters anything below the glomerular cut-off, so the same small size that aids diffusion also ensures urinary loss. What is not filtered is captured by reticulo-endothelial macrophages that detect anionic phosphate density as a damage-associated pattern; once opsonized, the duplex is trafficked to lysosomal degradation along with microbial RNA. Endothelial tight junctions present another lattice: even in tumors with aberrant vasculature, pores are intermittent and heterogeneously distributed, so transcytosis becomes a stochastic lottery rather than a reliable outcome. If a nanoparticle can make it to the abluminal space, it still faces an extracellular matrix that is rich in glycosaminoglycans that bind anionic cargo with millimolar affinity, setting up a local sink that reverses outward diffusion. Cellular uptake by endocytosis is deceptively encouraging, because most vesicles mature into acidified compartments where siRNA is sequestered and eventually shredded. Escape requires a fusogenic or proton-sponge motif, but the same chemistry that destabilizes endosomes can also leak into plasma membranes, tipping the therapeutic index toward toxicity. Finally, the cytosol itself is not a neutral playground; it is crowded with RNA-binding proteins that can sequester the guide strand away from Argonaute or, conversely, load the wrong strand and amplify off-target effects. Each of these barriers is individually surmountable, but their sequential multiplication collapses the overall probability, so that the fraction of injected dose that ultimately engages the RNAi machinery can be orders of magnitude below what is needed for pharmacological activity.
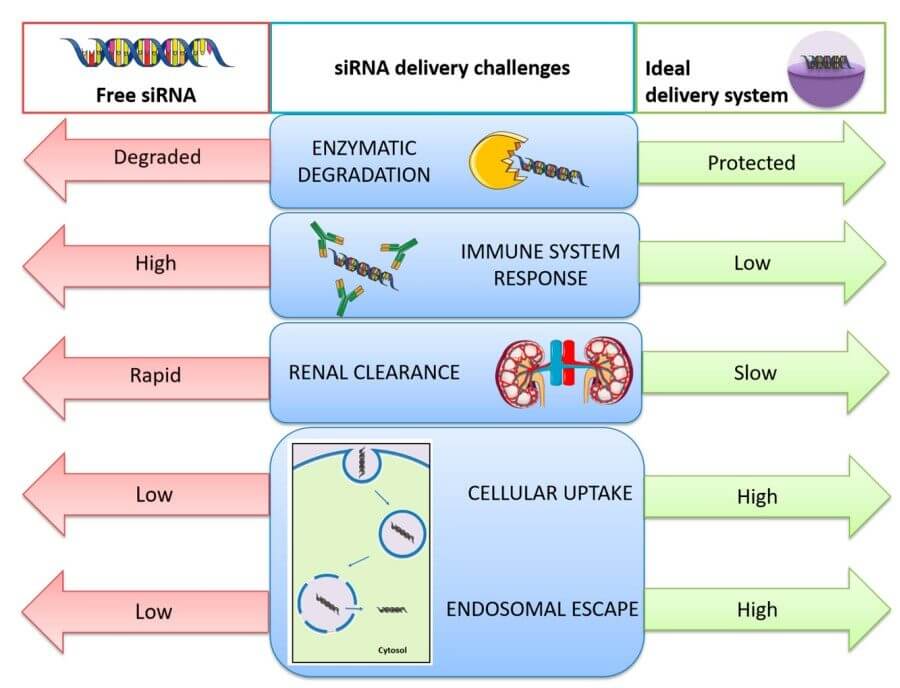 Fig. 1 Challenges related with in vivo small interfering RNA (siRNA) delivery and solutions provided by the use of nanocarriers to overcome the problems of free siRNA delivery.1,5
Fig. 1 Challenges related with in vivo small interfering RNA (siRNA) delivery and solutions provided by the use of nanocarriers to overcome the problems of free siRNA delivery.1,5
siRNA does not fail in vivo because the logic of gene silencing is flawed, but because each physiological corridor between syringe and cytosol is lined with nucleases, charge-repulsive membranes, macrophage sentinels and acidified dead-ends. Each barrier is independently survivable, but their sequential multiplication collapses the probability that an intact guide strand will ever meet its complementary mRNA. The following sections anatomise the four most frequently cited failure modes—degradation, uptake blockade, endosomal entrapment and off-target toxicity—while emphasising that they are not discrete hurdles but mutually reinforcing liabilities that must be negotiated in unison.
Following removal of the infusion needle, the naked siRNA is immediately present in a highly ribonuclease-enriched environment, where all exogenous RNA is considered molecular trash. Pancreatic-type superfamily extracellular RNases have a cationic active site that binds the anionic phosphate backbone and catalytically hydrolyze the phosphoester bond at the 3′-O-P position within seconds. The 5'-to-3' polarity of duplex RNA is not protective; in fact it is detrimental as perfectly blunt ends are recognizable features of viral replication intermediates, which results in an even more rapid catalysis. Serum albumin and low-density lipoproteins can also act as co-factors for RNases by binding the substrate into a conformation that is ideal for nucleolytic processing. Renal filtration also becomes an issue as siRNA and other oligonucleotides below the glomerular filtration cut-off are swept into the ultrafiltrate. There, luminal RNases proceed to digest the oligonucleotide before it is excreted in the urine. While certain chemical modifications such as 2′-O-methylation or phosphorothioate linkages may slow down these processes, it is not possible to prevent degradation entirely as the modifications leave a scissile phosphoester bond at each terminus of the oligonucleotide. Furthermore, the higher degree of backbone modification results in increased hydrophobicity which leads to adsorption to the vessel wall. The vessel wall adsorption forms a secondary sink for the molecules, effectively clearing it from circulation. Degradation of the intact siRNA is therefore a distributed process that begins in the blood, continues during glomerular filtration and ends in the urine, resulting in a very small fraction of the administered dose arriving intact at the target cell membrane.
The initial barrier to translocation is the plasma membrane, an electrically polarized lipid raft whose outer leaflet is studded with a glycocalyx of sialylated proteoglycans. The encounter with this structure represents an electrostatic cliff, with the net negative charge of the phosphate backbone repelled by the anionic sulphate and carboxyl groups that festoon the heparan sulphate chains. Passive diffusion across this barrier is thermodynamically forbidden because the hydrophilic siRNA cannot partition into the bilayer core, and receptor-mediated pathways are not an option because they demand multivalent interactions that cannot be sustained by a single 13 kDa duplex. In endocytosis, which is the most frequent mode of uptake, the trajectory is usually non-productive. Clathrin-coated pits internalize the molecule into early endosomes that mature into late compartments whose luminal pH decreases to 5.5, protonating histidine residues and activating nucleases that pre-empt silencing. Macropinocytosis is an alternative portal, but the resulting vacuoles collapse within minutes, trapping cargo in a cortical rim from which escape is improbable. Even forced uptake with cationic carriers offers an illusory gain, because electrostatic condensation often drives the particle into recycling endosomes that circle back to the membrane and eject their contents extracellularly. Inefficient uptake is thus not merely a quantitative shortfall, but a qualitative misrouting that sequesters siRNA in sub-cellular depots where it is either degraded or expelled before cytosolic delivery can occur.
The most commonly reported but least often circumvented hurdle to siRNA delivery is endosomal entrapment. Vesicles that have been internalised initiate a maturation programme regulated by Rab GTPases, which bring in proton pumps and chloride channels that gradually acidify the lumen while concentrating lysosomal hydrolases. siRNA cargo that stays in this transport is likely to be degraded, as the half-life of RNA at pH 5 is many times lower than at cytosolic pH. Membrane fusion peptides are chronically challenged by a timing paradox: a fusogen must be inactivated in the bloodstream to prevent complement activation but must be lytic in the small pH range between early and late endosomes. Proton-sponge polymers try to use this gradient to draw in protons until osmotic swelling lyses the vesicle, but the same cationic buffering that is used to 'spring' the transfection reagent also buffers cytosolic pH, indirectly activating stress kinases that phosphorylate translation factors to inhibit protein synthesis. Photochemical or redox-cleavable lipids can be used to bring more spatial control, but depend on exogenous triggers (light or reactive oxygen species) which rarely have uniform tissue penetration. Even if physical escape is obtained, the abrupt change in pH can denature the siRNA duplex, separating the guide and passenger strands and permitting the passenger strand to be loaded into Argonaute. In this sense, poor endosomal escape is not merely the failure to leave an organelle, it is a series of kinetic, chemical, and biological limitations that must be aligned within a few minutes to avoid lysosomal degradation and cytosolic mis-loading.
Off-target effects are the consequence of RNAi's main technical appeal: its catalytic efficiency. A single guide strand can cycle through dozens of mRNAs, so low-probability mismatches can generate phenotypic noise. Seed-region complementarity of six bases is enough to seed Argonaute recruitment if the "off-target" transcript is abundant. But because this transcriptional interference is sequence-specific, it is invisible to routine safety panels, which only detect cytotoxicity or cytokine release. Immunogenicity is another mode of off-targeting: GU-rich sequences or blunt ends can bind endosomal Toll-like receptors (TLRs), unleashing a type-I interferon response. The subsequent "off-target" gene-silencing is actually a non-specific antiviral programme. Chemical groups that quench TLR activity increase lipophilicity, causing hepatocyte or splenic macrophage uptake and steatosis or splenomegaly in repeat-dose studies. More insidiously, the endogenous microRNA pathway can be saturated if the siRNA competes for Exportin-5 or Argonaute loading, with consequences like repression of endogenous microRNAs and de-repression of oncogenes that were being held in check by those microRNAs. Off-target effects thus present not just a side-effect but a systems-wide disruption that can take weeks to develop and which requires transcriptomic monitoring long after traditional toxicology assays.
Nanoparticle technologies have shifted the siRNA delivery paradigm from a single molecule game of chance to a systems level interaction with the host milieu. By jointly designing the parameters of size, surface chemistry and release kinetics, these vectors establish a "nano-shield" microenvironment for the duplex while presenting it with physiologic cues to drive it across membranes, through endosomes and into the cytosol without setting off innate alarms. The following sections describe how three classes of vectors – lipid, polymeric and ligand-decorated carriers-operationalize this conceptual shield-and-guide strategy through specific structural decisions.
LNPs succeed in part because they are written in the same lipid grammar that cells have evolved to traffic their own contents. A cationic or ionisable amphiphile first electrostatically condenses the duplex into a 100-nm core coated by distearoyl phosphatidylcholine and cholesterol; the same lipid that confers stability to viral envelopes now shields siRNA from ribonucleases and complement. PEGylated lipids introduced at a few mole-percent form a steric brush that resists opsonisation, yet are anticipated to desorb after the first hour to unmask underlying cationic charge for membrane apposition. The innovation here is the pH-sensitive amphiphile whose apparent pKa is just below extracellular pH: at 7.4 the surface is almost neutral to minimize non-specific interactions, but in the early endosome the fall to 6.0 protonates the head-group, driving electrostatic destabilization and flip-flop of anionic lipids from the cytoplasmic leaflet. This transient pore permits siRNA to leak out before the vesicle matures into a degradative lysosome. Recent iterations feature secondary fusogenic phosphatidylethanolamine derivatives that lower the activation energy for membrane rupture, yet the same chemistry risks haemolysis if the particle inadvertently ends up in erythrocytes. To obviate this, the outer monolayer is doped with a zwitterionic ceramide that increases the bending modulus to deflect the carrier toward hepatocytes, where sinusoidal fenestrae facilitate direct contact. The result is a self-editing vector that is innocuous while in circulation but becomes lytic only inside the intended intracellular compartment, reconciling potency with tolerability without resorting to exotic materials.
Polyplexes leverage the statistical avidity of many cationic repeat units instead of the unitary charge on a single lipid. The linear or branched polymer winds the siRNA into a 50–80 nm toroid that has sufficient positive surface potential to drive adsorptive endocytosis, but the same charge density will kill the cell if it aggregates on the mitochondrial surface. To avoid this, modern vectors are constructed with proton-sponge motifs, tertiary or histidyl residues whose stepwise pKa ladder functions as a buffering unit between pH 7.4 down to 5.0. For each proton imported, a chloride counter-ion osmotically swells the endosome until its physical rupture causes wholesale release of the polyplex. The polymer backbone is also designed to hydrolyze or unlink by disulfide cleavage once cystolic glutathione has cleaved the backbone, so that excess cationic charge does not linger to disrupt the inner mitochondrial leaflet. The kinetics can be optimized by mixing a high-molecular-weight fraction for compaction with a short oligomer that functions as a charge quencher to delay the onset of membrane disruption until late endosomal arrival. As synthesis is iterative, other domains like nuclear localization signals, stealth PEG, or collagen-mimetic peptides can be appended without reformulating the entire particle. The resulting construct acts like a temporal bomb whose fuse is lit by acidification and snuffed by cytosolic reduction, creating a chemically flexible alternative to lipid systems with an equivalent safety margin.
Ligand-mediated targeting effectively changes the role of the nanoparticle from a stochastic blood-borne entity to a ligand–receptor puzzle piece. The small molecule, peptide or single-chain antibody is conjugated using hetero-bifunctional linkers that are carefully chosen to maintain binding affinity and colloidal stability. The ligand density must be below the threshold where receptor cross-linking and subsequent lysosomal sequestration occurs but above the density needed to compete with endogenous ligands. Modelling suggests a few hundred ligands per particle may provide the best balance, but the real vasculature is heterogeneous. The shear stress in inflamed capillaries may strip weaker conjugates, while basement-membrane glycans present a steric jungle that buries the recognition motif. To overcome these variables, flexible PEG tethers can be introduced to extend the ligand beyond the PEG brush so that it can "fish" for receptors even when the particle itself is sterically hindered. Dual-targeting strategies can also refine specificity: a primary ligand that binds a constitutive receptor can be used for initial tethering to the cell surface, and a secondary pH-sensitive peptide can be exposed in the acidifying endosome to bind a different epitope and reroute the vesicle towards a recycling rather than a degradative pathway. The ideal construct is a logical AND gate, where uptake only occurs if both signatures (for example a tumor-specific integrin and a low-pH microenvironment) are present, minimizing on-target but off-tissue toxicity. Molecular programming of this kind turns the nanoparticle into a context-aware vehicle that discriminates between physiological and pathological milieu in the absence of external triggers.
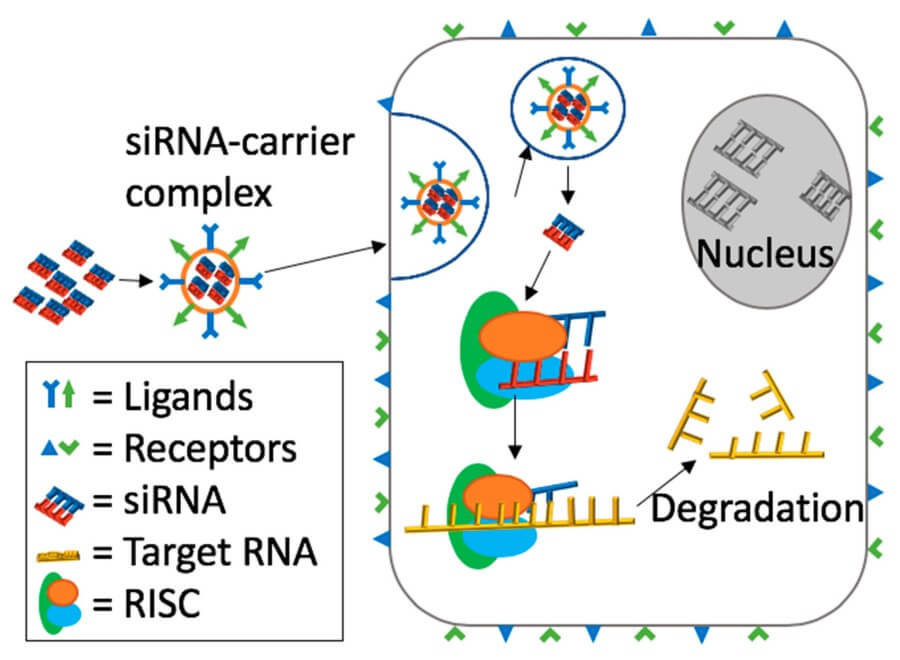 Fig. 2 General mechanism of ligand-receptor mediated targeting and siRNA delivery.2,5
Fig. 2 General mechanism of ligand-receptor mediated targeting and siRNA delivery.2,5
Proof-of-concept delivery programmes in real patients have now been reported for organs once thought inaccessible to systemic RNA. Here, we present a selection of case studies that exemplify how the combination of nanoparticle design, administration route and disease biology can result in clinically actionable gene silencing.
Initial ionizable-lipid particles were developed for hepatocyte targeting, due to the abundance of fenestrated endothelium and apolipoprotein E ligands in the liver. A single intravenous injection of transthyretin-targeting siRNA led to persistent knockdown of up to a year, demonstrating the potential for durable RNAi and laying the foundation for a platform that could be tailored to other liver-specific transcripts including PCSK9, factor XI or glycolytic enzymes. Subsequently, researchers modified this platform by substituting the E-apolipoprotein–targeting lipid with a more hydrophilic version, and re-targeting to Kupffer cells rather than hepatocytes, to potently silence inflammatory cytokines without changing the nucleotide sequence. This macrophage tropism is now being exploited in indications with a fibrotic or infectious component where resident phagocytes are implicated in pathogenesis.
Polymer nanocarriers coated with polysorbate-80 were used to shuttle siRNA targeting myostatin across the blood–muscle barrier in a murine model of muscular dystrophy. Repeated intravenous injections on a weekly basis led to persistent knockdown, increased fiber diameter and enhanced grip strength without increases in liver enzymes. The key feature was a redox-cleavable disulfide in the polyester backbone: high extracellular glutathione maintained particle integrity, whereas the lower cytosolic redox potential led to depolymerization and rapid release of siRNA. A follow-up study used the same platform to silence mutant repeat transcripts in myotonic dystrophy, and demonstrated that nanoparticle tropism could be tuned simply by changing the hydrophilic-lipophilic balance of the corona block rather than by re-engineering the RNA payload.
Intravitreal injection of hyaluronan-coated polymer micelles was able to mediate uptake of siRNA into the retinal pigment epithelium and silence VEGF-A, resulting in the suppression of neovascularization in a model of laser-induced choroidal neovascularization. The hyaluronan coating bound CD44 receptors, which are overexpressed on inflamed endothelium, providing dual retention and cell-specific uptake mechanisms. The lack of macrophages in the vitreous humor extended the half-life of particles to weeks, allowing for quarterly dosing intervals in line with the schedules of currently used anti-VEGF antibodies. A follow-on study showed that this same particle could be delivered through suprachoroidal injection and induce gene silencing in the posterior segment without puncturing the lens–iris barrier and causing cataracts.
At BOC Sciences, we specialize in overcoming the complex barriers of siRNA delivery through advanced nanoparticle engineering and customized formulation development. Our mission is to help biopharma and biotech partners accelerate RNA-based drug discovery and development with reliable, scalable, and clinically viable delivery solutions.
We provide end-to-end siRNA delivery services designed to transform early-stage ideas into validated therapeutic candidates.
Our core expertise includes:
Partnering with us means gaining access to a team of RNA delivery specialists and state-of-the-art nanoparticle platforms. We bring together deep expertise in lipid chemistry, polymer science, and bioanalytical validation, providing solutions tailored to your specific therapeutic target and development phase.
Our advantages include:
With a strong track record in customized nanoparticle design and siRNA delivery optimization, we enable our partners to achieve superior gene silencing efficiency, safety, and translational potential. Whether you're exploring oncology, rare disease, or genetic disorder pipelines, our delivery systems are designed to meet your most demanding R&D and clinical goals. At BOC Sciences, we don't just provide technology-we provide a partnership for success in siRNA delivery. Contact us today to discuss your project and discover how our platforms can accelerate your next RNA therapeutic breakthrough.
1. What are the main reasons siRNA delivery fails in research or clinical settings?
siRNA delivery often fails due to rapid degradation by nucleases, inefficient cellular uptake, poor endosomal escape, and off-target effects. These factors significantly reduce therapeutic efficacy.
2. How do nanoparticle systems improve siRNA delivery success rates?
Advanced nanoparticles protect siRNA from degradation, enhance cellular uptake, and promote endosomal escape, leading to more consistent gene silencing outcomes.
3. Which types of nanoparticles are most effective for siRNA delivery?
Lipid nanoparticles (LNPs), polymeric nanoparticles, and ligand-targeted systems have shown the highest efficiency and safety in both preclinical and clinical settings.
4. How can biotech companies optimize siRNA formulation?
Partnering with specialized siRNA delivery experts helps tailor nanoparticle composition, particle size, and targeting ligands to specific therapeutic needs.
References
 Loading ......
Loading ......