Pharmacology of tumors has changed conceptually over the past ten years, from a practice of poisoning fast-dividing cells to an effort to delete the molecular blueprints that endow immortality. Small-interfering RNA offers a means to achieve this goal directly, but the clinical development of siRNA in oncology has been less a tale of drug discovery than a quest for drug delivery. Initial systemic injections revealed that naked siRNA is metabolized within minutes, incites an innate immune response, and preferentially accumulates in the renal cortex rather than in the tumor stroma. The field has consequently re-directed its intellectual resources towards carrier designs—lipid nano-emulsions whose surface charge reverses at pH 6.5, polymeric vesicles that divest themselves of polyethylene glycol after extravasation, and exosome-mimetic vesicles whose CD47 ligands send a "do-not-eat" message to macrophages. These systems do not merely shield the duplex; they re-engineer its biodistribution, navigating it across endothelial fenestrations that only open around tumor masses and releasing it in response to redox potentials that are higher within the malignant cytosol. The fusion of such stimuli-responsive release with transcriptome-wide specificity algorithms has yielded a therapeutic index for siRNA that is comparable to that of small-molecule kinase inhibitors, breaching solid tumors that were previously deemed inaccessible to nucleic-acid therapeutics.
Mutations result in a syntax error in signalling networks; this includes a deletion of a stop codon to relieve growth restriction or insertion of an exon that encodes a protein that provides a survival advantage. siRNA acts as a molecular copy-editor by deleting the sentence that has been transcribed from the mutated or epigenetically amplified locus, without making any permanent changes to the genomic master copy. The implication for cancer therapeutics is that siRNA has utility beyond gene knock-down, as it is a programmable co-adjuvant that can be re-written for each new driver mutation, and so it liberates clinicians from the me-too clinical development treadmill that typifies standard chemotherapy. Since siRNA works at a post-transcriptional level, it can abrogate oncogenic isoforms created by alternative splicing or fusion that small molecules are sterically incapable of inhibiting. Furthermore, the same delivery vehicle can ferry multiple guide strands with different parameters, each strand can be aligned to the same driver pathway obliterating the redundancy that tumors use to outsmart single agents. The potential, therefore, is not in substitution for cytotoxic agents, but in recontextualizing their function: genomically matched siRNA cocktails silence the tumor cell anti-apoptotic gatekeepers and induce a biochemical remission that cytotoxics then fix as an irrecoverable death.
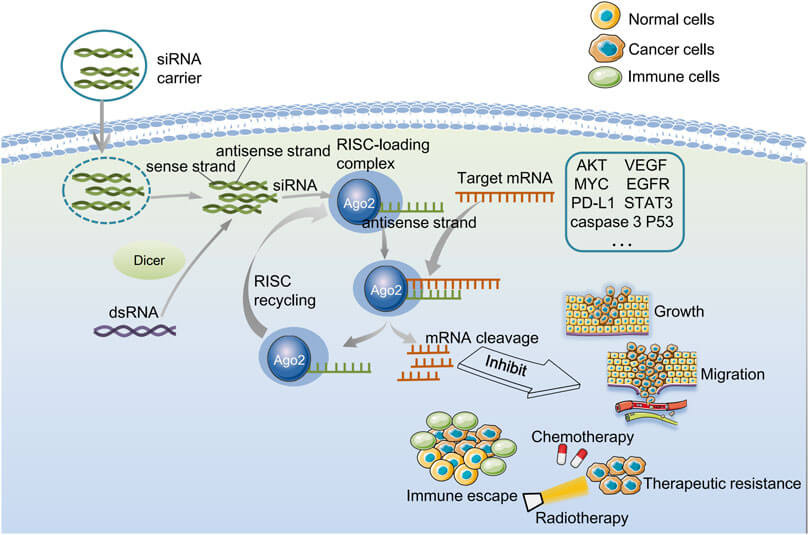 Fig. 1 Biological characterization of gene silenced by endogenous siRNA and exogenous siRNA.1,5
Fig. 1 Biological characterization of gene silenced by endogenous siRNA and exogenous siRNA.1,5
Oncogenes are usually not just over-expressed but over-heard, emitting autocrine growth signals that override normal quorum-sensing. Therefore siRNA silencing is needed to persist through the time needed for both oncogenic signalling and repressor titration, so that some redundant amplification can't restore the signal. Carriers have been developed to allow for this extended silencing, using polymeric micelles that erode slowly enough to release siRNA over more than one cell cycle and avoid the regrowth seen after a single siRNA pulse. In addition, 2′-fluoro modifications to the seed region (position 2–8) of siRNA increase the thermodynamic cost of mismatched base pairing, while phosphorothioate modifications to the 3′ end of siRNA confers exonuclease resistance to allow sufficient time for RISC to complete many rounds of catalysis. In the clinic this has been shown to affect tumors whose growth is dependent on point-mutated GTPases, where guide strands have been engineered to be allele-specific against a single-base mismatch, allowing silencing of the mutant transcript while leaving the wild-type allele intact for the essential normal stromal function, leading to tumour vascular collapse without the global fibrosis that caused problems with small-molecule GTPase inhibitors.
Drug resistance is a dynamic process, not a genetic mistake: cytotoxic selection pressures stimulate surviving clones to rewire their circuitry and up-regulate efflux pumps, DNA-damage bypass polymerases and anti-apoptotic chaperones. siRNA silences this conversation at the translational level, denying resistant cells the protein products required to survive chemotherapy. Newly designed carriers co-encapsulate siRNA targeting multidrug-resistance exporters together with a conventional cytotoxic agent inside the same nanoparticle, so both payloads arrive at the same intracellular destination and the temporal gap that allows for resistance to return is removed. The carrier itself can be tuned to the acidic microenvironment characterizing residual tumors after first-line therapy, releasing its cargo only where resistant clones are resident. Most elegantly, siRNA can be designed to target stemness transcription factors that are necessary for the maintenance of the slow-cycling reservoir that causes late relapse. The cocktail pushes these cells into cycle before chemotherapy, converting a dormant threat into a drug sensitive target. The clinical translation of these approaches is starting to result in prolonged complete responses in cancers that were otherwise characterized by nearly universal recurrence, recasting siRNA from a bench-top curiosity into the backbone of curative therapy.
In theory, siRNA should be able to target any oncogenic transcript. However, the stepwise journey from systemic injection to cytosolic RISC loading in vivo is fraught with physiologic obstacles, many of which are unique to malignancy. Tumours are not just masses of transformed cells; they are self-protective ecologies in which aberrant vasculature, dense extracellular meshes and immunosuppressive cocktails combine to sequester or degrade invading nanoparticles. The same features that underlie the "enhanced permeability and retention" effect also create elevated interstitial pressure, so that carriers extravasate with ease, but stall at the perivascular rim, never reaching the hypoxic cores where resistant clones survive. Further downstream, cancer cells are moving targets in their own right, shifting receptor profiles and endocytic rates within the same lesion so that a formulation that is well-optimized for one sub-population may be rejected by its neighbors. As such, delivery in oncology is less a single barrier, than a concatenated series of micro- and nano-scale hurdles that must be overcome in sequence; failure at any tier relegates the siRNA to an expensive placebo.
The tumor microenvironment is an abnormal organ with specific anatomy and physiology whose sole function is to resist drug delivery. Fenestrated endothelium allows nanoparticles to escape the bloodstream, but lack of functional lymphatics generates outward convection current that resist deeper penetration; as a result, siRNA accumulates within few cell diameters from vessels, sparing hypoxic niches. Dense and stiff fibrillar collagens, tenascin-C and hyaluronan make up a viscoelastic scaffold that sieves particles larger than a virus while cancer-associated fibroblasts contract the matrix, reducing interstitial space and increasing hydraulic pressure. Acidic catabolites drop the extracellular pH to the point where it protonates histidine-rich carriers, which causes premature release of siRNA in the stroma instead of inside tumour cells. Myeloid-derived suppressor cells secrete ribonucleases that degrade naked RNA, and macrophages phagocytose particles coated with opsonins, discarding the payload before it reaches malignant cells. Even if the carriers manage to escape these intruders, the abnormal vasculature is tortuous and arterio-venous shunts are frequent, so blood transit time is reduced and therefore the chance of wall contact is diminished. To overcome some of these barriers, next generation vehicles carry matrix-degrading enzymes tethered to the surface via acid-labile linkers, so that collagen is digested only after the particle senses the low tumor pH and transient channels are created that permit deeper diffusion. Others take advantage of the stiff matrix itself: rigid lipid polyhedra with aspect ratios designed to thread along collagen fibers migrate like bacteriophages through the mesh and are found delivering siRNA centimeters away from blood vessels in orthotopic models. As such, the microenvironment is not just a passive barrier but a hostile environment whose physical, chemical and cellular defenses must be overcome in a spatiotemporal manner if gene silencing is to take place in the right place at the right time.
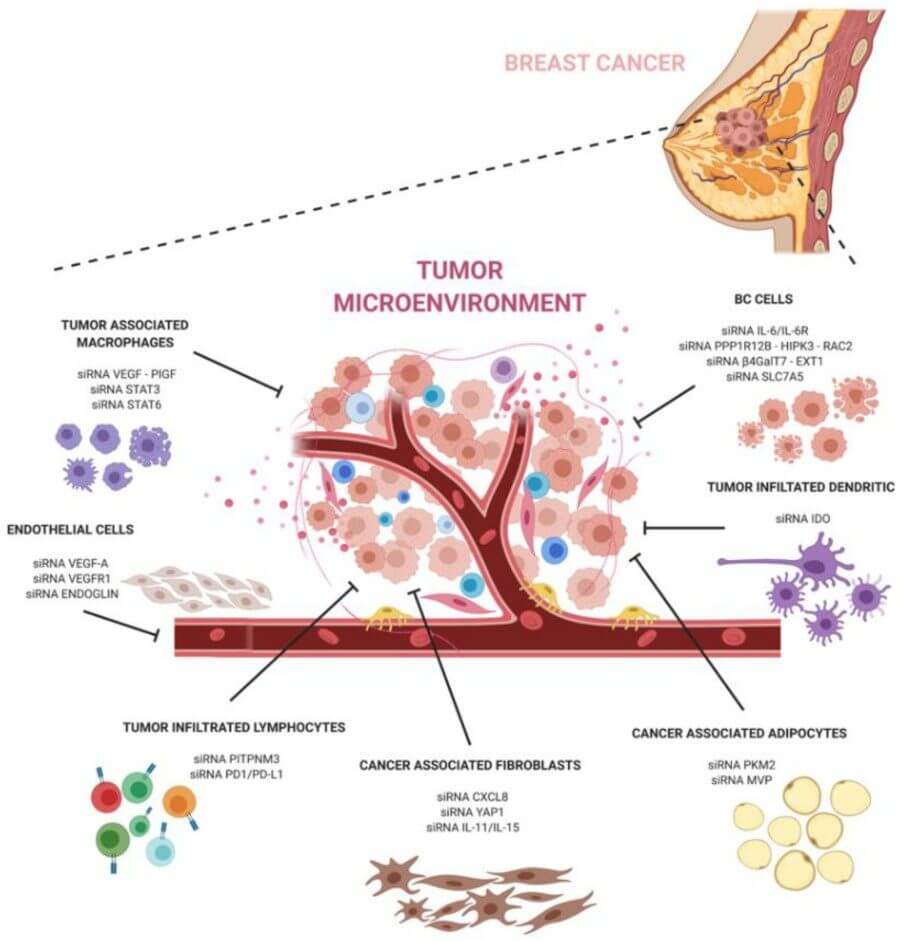 Fig. 2 A schematic representation of siRNAs against major components of the breast cancer microenvironment.2,5
Fig. 2 A schematic representation of siRNAs against major components of the breast cancer microenvironment.2,5
Intra-lesional heterogeneity ensures that a targeting approach may work one day but not the next as clonal hierarchies change. Genomic instability gives rise to polyclonal receptor expression: one sub-population over-expresses integrin αvβ3, another has a preference for CD44 variant isoforms, while a dormant clone down-regulates both and up-regulates caveolin-1 to change its major endocytic pathway from clathrin-mediated to caveolae-dependent. A ligand-targeted carrier therefore designed for the prevailing clone will by-pass minor reservoirs that go on to seed relapse. Epigenetic plasticity gives rise to a second layer of variability; transient methylation of the transferrin receptor promoter can silence its expression for days, long enough for the cell to escape a single-dose siRNA cycle. Mechanical heterogeneity is also important: dense cores have elevated cytosolic tension that squeezes endosomes, hampering their rupture and entrapping siRNA in degradative compartments, whereas loosely attached cells at the tumor invasive front undergo accelerated macropinocytosis, sequestering so much cargo that off-target effects appear. Temporal heterogeneity is similarly frustrating; the membrane's fluidity changes according to cell-cycle position, so the same nanoparticle will penetrate S-phase cells using electrostatic interactions, only to encounter G0 quiescent neighbors that push it away. Adaptive delivery systems overcome this moving target using bispecific ligands that can engage two receptors simultaneously, which reduces the likelihood that any single down-regulation event will permit escape. Others harness quorum-sensing peptides that stressed clones secrete, peptides that will bind to and activate a conformational switch on the particle, unmasking a cryptic cell-penetrating motif that allows entry even when the classical receptors are absent. The utilization of heterogeneity as an evolutionary battlefield rather than an unchangeable obstacle enables these strategies to convert tumour cell diversity from an unsolvable barrier into an engineering problem that can be addressed.
The discovery that cancer-causing messages can be erased in addition to being suppressed has moved small interfering RNA from the status of a research tool to that of a potential mainstay of anti-cancer medicine. However, tumour tissue is a complicated delivery landscape: abnormal blood vessels, high internal pressure and an immunosuppressive microenvironment work together to ensnare or break down arriving RNA. The focus of recent advances has thus shifted away from the nucleotide payload and towards redesigning the journey from blood vessel to cytoplasm. By combining ligand-targeted nanoparticles with natural carriers that can mimic the host's own molecules, and by timing the gene silencing to coincide with chemotherapy or metabolic stress, researchers are now able to reach tumour-to-liver ratios that would have been considered impossible just ten years ago. The result is that siRNA is no longer a vulnerable oligonucleotide but a programmable therapeutic whose effects can be focused on cancer cells alone, in a process that is redefining the therapeutic index of nucleic-acid medicines in cancer.
Traditional lipid bilayers passively extravasate and amass in tumor foci but they do not translocate beyond the perivascular rim, where interstitial pressure is maximal. The most advanced nanoparticles use a "programmed shedding" design: an outer stealth corona, which is repellant to opsonins, is sequentially lost under low pH, to unmask an intermediate layer of cell-penetrating peptides and tumor-specific ligands. In contrast to the simple cationic electrostatic lock, the inner core is formed by disulphide-cross-linked polymers, which are degraded by glutathione in the cytosol, allowing for siRNA release only in the Argonaute-containing compartment. To traverse the dense collagen scaffold, the carriers are formed into rod-shaped or polyhedral geometries with long axes oriented along fibrillar pathways, thus achieving a diffusion length that approaches that of viruses. The surface chemistry is also customized to target receptors that are over-expressed on malignant endothelial cells (e.g. integrin αvβ3 or neuropilin-1), so that the initial binding event takes place on the vasculature itself, thereby "pre-concentrating" the cargo before it ever reaches parenchymal cells. After being internalized, protonatable lipids in the particle then buffer the acidification of the endosome, thereby postponing lysosomal fusion and giving a wider temporal window for cytosolic release. The combined effect is a nanoparticle that functions as a self-steering missile: it homes in on the neoplastic postcode, it breaks through the stromal fortress and it then deposits its silencing cargo without activating the innate immune system or causing thrombocytopenia, toxicities which historically limited the clinical utility of earlier generations of cationic liposomes.
Ideal delivery vehicles would be endowed with the ability to transport nucleic acids between tissues without activating the immune response. To this end, researchers are taking advantage of the exosome's evolution as a traffic signal, endowing siRNA therapeutics with properties of self. Initially, stable producer cells are engineered to express a synthetic membrane protein that directs anchoring of a high-affinity tumor-targeting peptide (commonly Lamp2b) to the outer membrane at a density that is sufficient for avidity-based receptor binding on cancer cells but not so high as to be rapidly cleared by the liver. The RNA is loaded either by electroporation or, more recently, by co-transfection of a plasmid encoding RNA-binding domains that bind to a sequence motif and specifically bind to and enrich the guide strand during exosome biogenesis. As the vesicle retains the surface proteins of its parental cell membrane, including CD47 and PD-L1 fragments that send "do-not-eat" signals, it escapes phagocytes and has a prolonged half-life. Within tumor tissue, the exosome membrane fuses directly to the plasma membrane of the target cell, without the entrapment in the endosomes that limit synthetic liposomes. This fusion is favored by the acidic microenvironment, which destabilizes vesicular phospholipids to a more fluid phase and induces membrane curvature required for hemifusion intermediates. Exosomes also lack viral proteins that can activate toll-like receptors, allowing for repeated administration without the cytokine bursts that make adenoviral vectors difficult to deliver. The latest generation of these therapeutics have combined the ability to silence genes with the ability to intervene in metabolism, co-delivering a siRNA against KRAS with a glycolytic inhibitor so that oncogene inhibition is coupled with energy deprivation, thus enhancing apoptosis and limiting escape pathways. The cell-derived nature of the vesicle delivery is what makes this platform clinically attractive, as once a master cell bank has been qualified, the production of the vesicles can be scaled up in bioreactors, in serum-free conditions, yielding a standardized, off-the-shelf product, with a surface topology and payload stoichiometry that is reproducible batch-to-batch.
Rarely does a single-agent siRNA eradicate a clinically established tumor, as the malignant genome is wired with redundancy and the suppression of a given driver frequently licences a compensatory pathway. Breakthrough regimens therefore seek to sequence or co-package siRNA with other modalities that act on orthogonal vulnerabilities. A prototypical example pairs an siRNA targeting an anti-apoptotic transcript with low dose DNA-damaging agents: the genotoxic insult up-regulates pro-survival signalling and thus sensitizes the cell to the subsequent silencing of MCL-1 or XIAP. Nanoparticle choreography is critical: both payloads must reach the same cell in a time window narrow enough to prevent transcriptional rebound yet wide enough to avoid overlapping toxicities. This is achieved by embedding the siRNA within a pH-responsive core and adsorbing the small molecule onto an intermediate lipid shell whose dissolution is triggered by endosomal acidity so that gene silencing precedes cytotoxic peak by a few hours. Immune checkpoint blockade is a second, synergistic layer: siRNA-mediated knock-down of VEGF or TGF-β normalizes vasculature and reduces extracellular adenosine, enhancing T-cell infiltration so that subsequent PD-1 inhibition encounters an inflamed rather than immune-desert microenvironment. Metabolic priming is an emerging third axis: glycolytic inhibitors or glutaminase antagonists lower ATP levels and thus suppress the active efflux of siRNA from endosomes via ATP-dependent pumps. The clinical translation of these concepts is beginning to manifest in protocol designs in which siRNA is administered on day 1 to collapse defence pathways, with chemotherapy or antibody on day 2 when tumor cells are transcriptionally committed to apoptosis. Such calendar-based combinations convert cytostatic gene silencing into cytotoxic synergy, providing durable responses without the cumulative myelosuppression that characterizes traditional multidrug cocktails.
At BOC Sciences, we are redefining how siRNA therapies reach and impact cancer cells. Our advanced nanoparticle delivery technologies are designed to overcome the major barriers of oncology drug development - from tumor microenvironment resistance to off-target toxicity - enabling our partners to accelerate clinical translation and therapeutic efficacy.
Cancer therapy demands precision, and our delivery systems are built with that principle in mind. We specialize in:
Each formulation is customizable to your oncology program's requirements - whether targeting solid tumors, hematologic malignancies, or metastatic lesions.
We offer end-to-end siRNA delivery services tailored to oncology pipelines. Our integrated approach includes:
By managing every stage within one collaborative framework, we reduce R&D complexity and accelerate your path to clinical readiness.
Our team combines expertise in oncology biology, RNA chemistry, and formulation science, supported by extensive data from preclinical and translational projects. This multidisciplinary foundation enables us to deliver high-performance siRNA systems that enhance tumor penetration, minimize immune activation, and ensure therapeutic reproducibility.
Collaborating with BOC Sciences means accessing more than just technology - it means partnering with a team that shares your mission to bring next-generation cancer therapies to patients faster. We provide scientific depth, flexible collaboration models, and regulatory-ready documentation, giving biopharma teams the confidence to move forward with bold innovation. Partner with us to unlock the full potential of siRNA in oncology. Contact our experts today to discuss a custom delivery strategy for your cancer therapeutic pipeline.
1. How does siRNA delivery contribute to cancer therapy?
siRNA can silence oncogenes and drug-resistance genes, enabling more targeted and less toxic cancer treatments.
2. What makes siRNA delivery challenging in oncology?
Tumor microenvironments, poor vascular access, and heterogeneous cancer cell populations hinder efficient delivery and gene silencing.
3. What are the latest breakthroughs in siRNA oncology delivery?
Tumor-targeting nanoparticles, exosome-based carriers, and combination therapies with chemotherapy or immunotherapy are leading innovations.
4. Are siRNA-based cancer therapies in clinical trials?
Yes—multiple siRNA oncology candidates are in advanced clinical phases, showcasing promising efficacy in solid and hematologic tumors.
References
 Loading ......
Loading ......