Hard-to-target cells don't just cloak themselves in a second lipid bilayer, they also reorganize their glycocalyx, fortify junctions, summon phagocytic body-guards and expel anything that "smells" foreign, long before a single siRNA phosphate can be de-protonated. The successful strategies are therefore not incrementally superior particles but platform programs that approach anatomy, immunology and PK as co-variables. By pairing chemical camouflage to ligand-mediated first contact, biopharma can now broker entry into compartments once deemed unreachable, turning cytosolic gene silencing from a hepatic oddity into a modular, organ-agnostic modality.
Hard-to-reach cells all have three primary barriers: limited fenestration of the vasculature, low endocytic activity and unfavorable environments. Fenestrations in endothelial tight junctions are small, as a result, particles larger than 20 nm cannot enter. Serum opsonins are abundant and promote rapid phagocytic clearance. In the event that a nanoparticle can extravasate, surface glycocalyces are anionic and repel anionic siRNA and endosomal trafficking preferentially routes vesicles toward lysosomal degradation instead of cytosolic release. As a result, it is not enough to develop delivery strategies that are optimized for size or tuned for charge; they must also be specific to cell type and include real time escape triggers that can sense pH, redox, or enzymatic changes in the microenvironment.
Immune cells are professional sentry cells, trained to sense anionic, double-stranded or blunt-ended RNA with pattern-recognition receptors connected to cytokine amplifiers. Coating siRNA in standard cationic liposomes therefore risks uptake by plasmacytoid dendritic cells and subsequent interferon cascades presenting as "off-target toxicity" but actually on-target innate activation. Efficient targeting therefore necessitates a 2-step stealth-and-engage choreography: a high density, neutrally-charged corona, typically zwitterionic phosphorylcholine or short-chain poly(hydroxyethyl acrylate), is added at a grafting density high enough to mask phosphate charge but not so high as to over-rigidify the particle and impede endosomal escape. A lineage-restricted ligand is then conjugated, e.g., an antibody fragment binding a receptor that is recycled but not signalling-competent, by a cleavable maleimide-thiol bridge. The ligand density is adjusted so that each particle presents fewer than a critical number of binding motifs and hence is unable to cross-link the target receptor and so route the carrier for lysosomal degradation. Upon entry, disulfide reduction in the glutathione-rich cytosol cleaves the ligand, releasing the siRNA to load RISC and the polymer scaffold to hydrolyse into renal filtration-sized fragments. This transient "masked-cationic" approach has allowed knock-down of pro-inflammatory kinases in macrophages without affecting whole blood cytokine profiles, demonstrating that immune cells can be silenced instead of activated when the delivery vector plays by their surveillance rules.
Neurons, however, provide an additional topological constraint: while the axon can extend for centimeters away from the soma, the RISC resides in the cytoplasm throughout. Simple somatic transfection will therefore leave distal mRNAs untouched. Convection enhanced delivery offers another way to bypass the BBB, but it also forms a fluid cavity that collapses within hours. Trapped particles end up in a perivascular rim. A more neuron-compatible vector couples the siRNA to cholesterol using a photocleavable spacer. The lipophilic appendix allows it to associate with high density lipoprotein particles which are internalised into neurons using endogenous lipoprotein receptors. Because cholesterol is trafficked retrogradely inside endosomes that are transported on microtubule rails, the duplex can now hitch a ride all the way back to the soma while being protected from nucleases. Illumination with near-infrared light through a thinned skull window then cleaves the spacer, releasing the siRNA into the cytosol exactly where RISC concentration is highest. The same lipoprotein shuttle can also be engineered to carry an oxime bond that is cleaved by the oxidative burst present in damaged axons, endowing the system with disease-site specificity without the need for stereotactic surgery. Early validation demonstrates knock-down of axonal tau mRNA throughout the entire cortico-spinal tract after a single intracisternal injection, showing that neurons can be accessed globally, if the vector piggy-backs on endogenous lipid trafficking highways, instead of trying to force a new entry portal.
Liver dominance owes less to inherent nanoparticle tropism than to vascular phenotype: sinusoidal fenestrae are open to anything below 100 nm, while the continuous endothelium elsewhere in the body requires active transcytosis for passage. To target kidney, lung or pancreas, carriers first need to escape the reticulo-endothelial filter and then need to overcome an organ-specific endothelial barrier. A validated strategy is to use an apelin-mimetic peptide that targets APJ receptors selectively expressed on fenestrated peritubular capillaries of the kidney; the particles are retained in the interstitium and are then taken up by proximal tubular cells that overexpress RISC. Pulmonary carriers exploit the fact that alveolar capillaries are lined by a basement membrane that is rich in heparan sulphate; the inclusion of a low-affinity, high-avidity heparin-binding domain in the PEG corona transiently tethers the carrier long enough for it to transcytose, via albumin-mediated pathways, into the airway lining fluid. Pancreatic islets are surrounded by a double basement membrane and supplied by micro-vessels that are devoid of fenestrae, so a two-wave strategy is needed: first a mild ultrasound pulse loosens the tight junctions for minutes and then nanoparticles with a neutral surface bearing a GLP-1 analogue bind to incretin receptors expressed on β-cells and undergo receptor-mediated transcytosis. The common denominator for all three organs is to regard the endothelium as a programmable checkpoint rather than an inert fence; by tuning ligand affinity to receptor recycling kinetics and by adjusting particle elasticity to vascular shear, extrahepatic deposition becomes a reproducible unit operation rather than a lucky accident.
Classic siRNA carriers – first-gen liposomes, polycationic dendrimers, and simple conjugates – were developed to address one problem at a time: The primary design of first-generation siRNA carriers included either packaging the duplex inside their structure or altering its charge density and applying a stealth surface layer. The barriers, in vivo, are multiplicative, not additive. A cationic liposome that shields RNA from serum RNase also opsonizes the particle, flagging it for Kupffer cells; a PEGylated micelle that dodges phagocytes at the same time stifles endosomal escape; a cholesterol conjugate that increases membrane affinity at the same time commandeers LDL receptors in off-target organs. Lacking feedback loops that would allow the carrier to remodel itself after passing each biological checkpoint, the same chemical feature that enables step A disables step B, turning the delivery pathway into a zero-sum game.
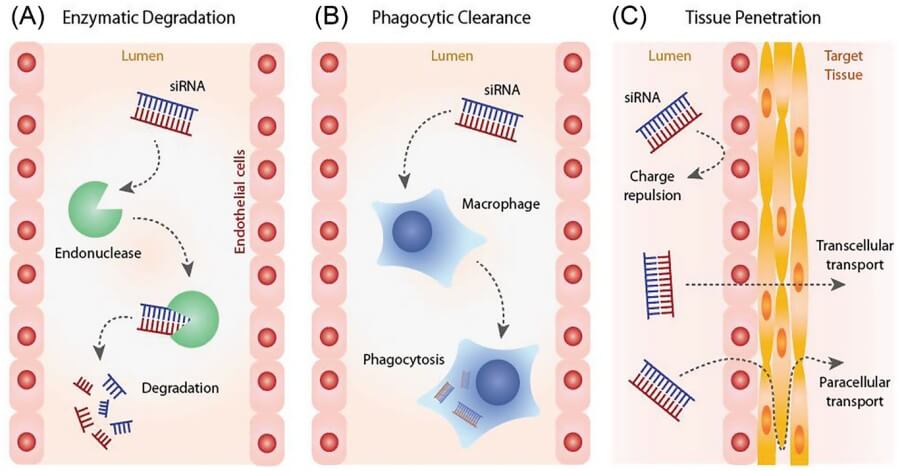 Fig. 1 Extracellular barriers to RNAi therapy.1,5
Fig. 1 Extracellular barriers to RNAi therapy.1,5
Conventional polycationic carriers were initially lauded for electrostatically condensing siRNA into sub-100 nm particles. The same amine density that compacts the duplex also protonates at physiological pH, transforming the complex into a membrane-disruptive detergent. This lytic potential is not limited to the endosomal leaflet: residual particles that accumulate at the mitochondrial outer membrane promote cytochrome c leakage and activation of intrinsic apoptosis cascades, masquerading as "siRNA off-target toxicity" but in fact carrier-induced necrosis. Moreover, the repetitive dosing needed for chronic silencing transforms the particle into a hapten: cationic lipids form adducts with serum albumin, generating neo-antigens that drive IgM and, on re-dosing, IgG responses. Antibodies function beyond drug clearance as they cross-react with natural lipoproteins to create temporary complementopathy which appears as infusion-related chest tightness and hypotension. The metabolites of biodegradable lipids (primarily acyl-choline derivatives) activate Toll-like receptor 4 on macrophages which initiates a cytokine storm that overwhelms RNA's sequence-specific immunostimulation. The Achilles heel of first-generation carriers, then, is not inefficacy but over-activity: they solve the membrane penetration problem by unleashing a chemical bulldozer that cannot distinguish between the endosomal bilayer and every other membrane it encounters.
Traditional complexation methods rely on bulk pipetting of siRNA into cationic lipids or polymers while mixing vigorously, which results in a stochastic distribution of core-to-shell ratio, surface potentials and lamellarities. As the biological fate of a nanoparticle is exquisitely sensitive to these parameters, where zeta potential shifts of a few millivolts can reroute clearance from renal to hepatic pathways, minor variations in temperature, ionic strength or injection rate translate into macroscopic changes in biodistribution. Regulatory agencies now demand that each manufacturing lot demonstrate equivalent knock-down in a surrogate tissue, yet the same polydispersity that governs efficacy also governs toxicity: a sub-population of oversized vesicles may be more efficient at endosomal escape but simultaneously more prone to complement activation. Attempts to tighten the specifications by post-production extrusion or dialysis inadvertently shear the particle, exposing fresh cationic patches that retrigger opsonization. Consequently, late-stage clinical trials often oscillate between under-dosing (no efficacy) and over-dosing (immune events), not because the therapeutic index is intrinsically narrow, but because the product itself is an ensemble of microscopically different vectors whose mean properties drift between batches. This reproducibility gap has become the implicit reason why several programmes advanced into phase II only to fail when scaled to commercial manufacturing lines, revealing that conventional complexation chemistry is incompatible with the statistical process control demanded of modern biologics.
In contrast to previous generations of delivery approaches, the latest generation of strategies no longer considers siRNA as a vulnerable payload that needs to be encased and shipped with hope and prayer. It develops vectors in which each chemical transformation is linked to a specific biological gate. Integrating ligand-receptor recognition, cell penetrating peptides and stimuli-responsive release modules, these systems transform the random uptake of first generation nanoparticles into a digital process that can be engineered on demand for each organ, tumor microenvironment and even intracellular compartment.
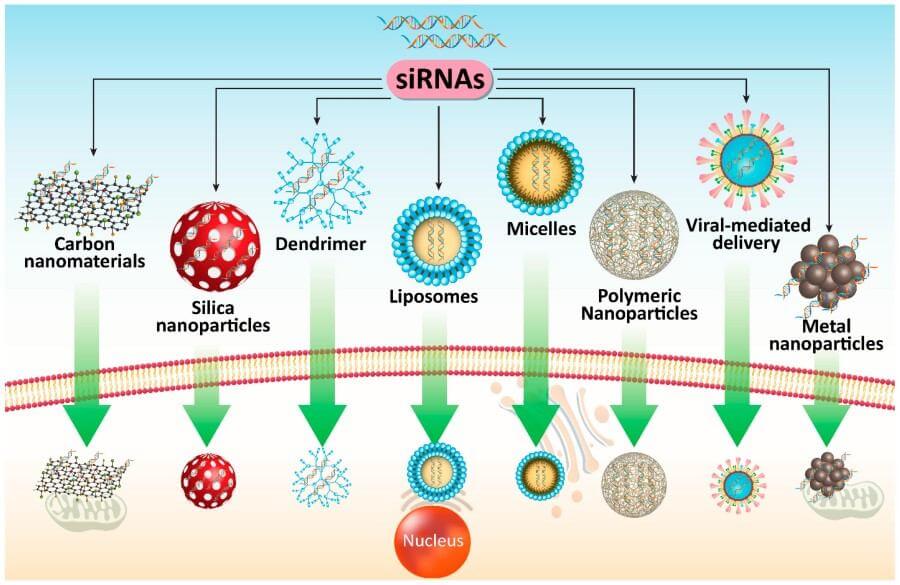 Fig. 2 Different co-delivery systems for siRNA in PC therapy.2,5
Fig. 2 Different co-delivery systems for siRNA in PC therapy.2,5
Ligand-mediated targeting instead pre-scribes a new biodistribution prior to injection. Small molecules, antibody fragments or high-affinity peptides are conjugated through hetero-bifunctional linkers whose spacer length is chosen to project the ligand beyond the PEG cloud, allowing it to "fish" for cognate receptors without being sterically masked. The choice of receptor is itself a pharmacokinetic decision: targets that recycle rapidly (e.g., folate or integrin receptors) afford multiple opportunities for uptake per vascular pass, whereas signalling-competent receptors (e.g., EGFR) can trigger internalization routes that bypass late endosomes altogether. Critically, ligand density is kept below the threshold that would cross-link receptors and provoke lysosomal retention; instead, a sub-stoichiometric surface coverage favors single-point attachment followed by clathrin-independent entry into mildly acid compartments. The same ligand–receptor pair is then used in reverse once inside: the receptor recycles to the membrane and the particle is stranded in a sorting endosome whose pH has not yet risen high enough to activate nucleases. This lag in time gives a kinetic window for secondary escape modules (fusogenic lipids or proton-sponge polymers) to function before degradation begins. The clinical benefit is an increase in the therapeutic index: because uptake is receptor-gated rather than charge-driven, doses can be reduced without loss of gene-silencing depth, lowering cost and risk of immunogenicity.
Cell-penetrating peptides (CPPs) provide a second mechanistically distinct portal: instead of passively waiting for a receptor to pull them in, they transiently disorder the lipid bilayer itself. Arginine-rich sequences in particular adopt a bidentate motif where guanidinium groups form bidentate hydrogen bonds with phosphate head-groups, creating local negative curvature that nucleates a toroidal pore. The key is then to switch this pore on only after the particle has docked onto the target cell; otherwise, it causes systemic membrane damage. Recent designs therefore mask CPPs with acid-labile amides or reducible disulfides that are cleaved in the early endosome, restoring the cationic charge exactly where membrane disruption is beneficial. To further restrict activity, CPPs are grafted as short "tails" on an otherwise neutral dendrimer; the tail length is tuned so that the pore lifetime is shorter than the diffusion time needed to reach off-target cells, confining lytic activity to the first membrane encountered. When combined with a primary targeting ligand, the CPP functions as a secondary key: receptor binding concentrates the particle, then endosomal acidification unlocks the membrane-penetrating module, releasing siRNA next to the cytosolic face. This dual-key logic has enabled neuron-specific knock-down after intravenous injection, a feat unattainable with either ligand or CPP alone.
Controlled-release nanoparticles consider the intracellular space as a series of pH, redox or enzymatic switches that can be wired in the material itself. A canonical example is an ionizable lipid whose apparent pKa is midway between plasma and endosomal pH: the core is anionic during circulation, minimizing complement activation, but becomes cationic inside acidifying vesicles, destabilizing the bilayer and releasing the duplex. The same particle can be further encoded with a disulfide cross-linker that dissolves in the cytosolic glutathione milieu, ensuring that any payload still trapped in residual endosomes is liberated minutes later. By embedding these triggers in a block-copolymer shell, release kinetics can be stretched from seconds to hours, matching the time needed for the guide strand to locate and load into RISC. Because the switches are reversible, the particle can even respond to repeated pH oscillations, offering multiple opportunities for escape if the first attempt fails. This fail-safe architecture has turned the notoriously inefficient process of endosomal exit into a probabilistic multi-draw lottery, raising cytosolic bioavailability without increasing peripheral exposure.
Rather than building carriers from scratch, extracellular-vesicle (EV) camouflage repurposes nature's own delivery service. siRNA is loaded into donor cells, secreted inside authentic vesicles, and then harvested with a surface proteome identical to the parent cell. The resulting particles inherit the tropism of the source cell—neuron-derived EVs cross the blood–brain barrier, while muscle-derived EVs home to skeletal tissue. To enhance specificity, donor cells are engineered to display a targeting ligand on the vesicle surface; because the ligand is synthesized by cellular machinery, it is correctly folded and glycosylated, avoiding the orientation errors common to chemical conjugation. The vesicle membrane itself acts as a built-in fusion device: cholesterol-rich microdomains laterally reorganize in acidic endosomes, lowering the energy barrier for back-fusion and releasing siRNA directly into the cytosol. This lipid-only escape mechanism circumvents the need for synthetic fusogens, reducing toxicity while preserving the vesicle's ability to evade mononuclear phagocytes through CD47-mediated "don't-eat-me" signalling. Early comparative studies show that EV-mimetic carriers achieve neuronal gene silencing at doses orders of magnitude below conventional liposomes, validating camouflage as a clinically translatable strategy for hard-to-reach tissues.
To realize this promise, process architectures will need to support translation to industrial manufacturing by incorporating biology as a unit operation: state-of-the-art single-use perfusion bioreactors are delivering liter-scale siRNA-lipid cocktails with inline, continuous-flow microfluidics that tightly control nanoparticle size distributions with low batch-to-batch variation (±5 nm) within campaign. Inline dialysis loops exchange buffer with WFI in real time, removing process hold tanks that have historically been incubators for aggregate formation. Downstream, closed-system sterile filtration trains can include 0.2 µm membranes that are pre-qualified for nucleic acid adsorption and virus-removal nanofilters with a pore size selected to retain the hydrodynamic diameter of the siRNA-lipid complex and avoid shear-mediated disruption. Critically, the same skid can be used to support liver, tumor, or CNS indications by simply exchanging the targeting ligand on a pre-assembled PEG linker; this approach turns fixed capital equipment expenditure into a portfolio option rather than a single-product millstone. The current regulatory paradigm for submissions is beginning to accept multivariate models that correlate inline spectroscopic fingerprints with release specifications. This will allow a reduction in lot review times from weeks to days and facilitate alignment between manufacturing cadence and the rapid iteration cycles demanded by biotech investors. health-economic translation is engineered in from day one. Cost-of-goods models view lipid excipients as commodities, negotiate siRNA synthesis on a per-base fee that decreases with scale, and take credit for solvent recovery against carbon taxes that are already real in European markets. The resulting cost curve can intersect orphan-disease pricing floors at volumes achievable in a 200 L single-use bag and, simultaneously, the same process skeleton can be linearly scaled to multi-tonne feedstocks once blockbuster indications secure reimbursement. By integrating pharmacoeconomic breakpoints into the process design space, every technical milestone - size distribution, encapsulation efficiency, endotoxin clearance - also de-risks both clinical development and commercial viability, turning molecular ingenuity into a reimbursable therapy patients will list and can reach.
Delivering siRNA to hard-to-target cells demands more than conventional formulation expertise - it requires deep scientific insight, advanced engineering platforms, and proven translational experience. At BOC Sciences, we provide a partnership built on all three. Our goal is to empower biopharma innovators to achieve efficient, precise, and scalable siRNA delivery across even the most challenging therapeutic areas.
Our team combines expertise in nanoparticle design, cellular biology, and formulation chemistry to create delivery systems that reach where others cannot. We specialize in developing custom lipid nanoparticles (LNPs), polymeric carriers, and ligand-targeted formulations for immune cells, neurons, and other difficult-to-transfect tissues. By leveraging cutting-edge formulation tools and in vitro/in vivo validation models, we ensure every project achieves optimal uptake, stability, and gene silencing performance.
Choosing the right partner means gaining not only technology but also a collaborative development pathway. At BOC Sciences, we offer integrated siRNA delivery solutions that cover every stage of your program:
This end-to-end approach minimizes technical risk, shortens timelines, and ensures your therapeutic candidate progresses efficiently from discovery to development.
Our strength lies in collaboration and transparency. Every project we undertake is managed through open communication, shared data, and regulatory-aligned documentation - giving our partners full confidence in their delivery strategy. With a proven track record in extrahepatic siRNA delivery and hard-to-target applications, we deliver not just results but long-term value through scalable, validated technology platforms. At BOC Sciences, we don't just formulate siRNA - we enable precision delivery for the world's most complex targets. Contact our RNA delivery specialists today to explore how we can accelerate your next therapeutic program.
1. Why are certain cells considered hard to target with siRNA delivery?
Cells such as neurons, immune cells, and endothelial cells have unique membrane barriers and low uptake efficiency, making siRNA transport more complex.
2. What technologies improve delivery to hard-to-reach cells?
Targeted nanoparticles, ligand-receptor binding, and cell-penetrating peptides are among the leading solutions to enhance intracellular delivery.
3. How can delivery systems ensure specificity without toxicity?
By using receptor-targeted ligands and biocompatible carriers, siRNA can be delivered selectively to target cells while minimizing off-target effects and immune activation.
References
 Loading ......
Loading ......