The thymidine–uridine fork is the first and most deterministic decision in oligonucleotide design: a single methyl group at C-5 converts a flexible, immune-evasive ribonucleoside into a rigid, DNA-compatible building block, thereby pre-programming metabolic half-life, duplex geometry and manufacturing solvent without altering the amidite coupling cycle; recognizing this boundary allows developers to stream crystalline thymidine amidites into solid-phase DNA lines while reserving aqueous uridine triphosphates for enzymatic RNA transcription, thereby embedding both chemical identity and process identity into the same molecular scaffold before first-in-animal studies begin.
Table 1 Molecular and manufacturing dichotomy between T and U
| Feature | Thymidine (T) | Uridine (U) | Therapeutic read-out |
| C-5 substituent | Methyl | Proton | DNA vs RNA fidelity |
| Sugar pucker | C2'-endo (south) | C3'-endo (north) | Duplex rigidity |
| Innate sensor | Minimal TLR7/8 | RIG-I substrate | Immunogenicity dial |
| Process solvent | Organic | Aqueous | Chemistry route |
The thymidine/uridine choice is the first design gate in oligonucleotide therapeutics: thymidine confers DNA-like stability and RNase H recruitment, whereas uridine offers RNA-like flexibility and translational efficiency; mis-matching base to modality forces costly late-stage re-optimization of protecting groups, analytical methods and impurity fate-mapping.
Base choice is the first permanent structural lever: thymidine provides 2'-deoxyribose support to compose gapmer backbones that hijack RNase H1 to digest huntingtin or SMN2 transcripts, while uridine supplies cis-2',3'-diol chemistry to pre-organize siRNA guide strands for argonaute loading. Thymidine's 5-methyl also decreases the major-groove dielectric constant to improve protein binding and reduce renal filtration rates, and its absence in uridine enables faster base-flipping and enzyme-catalyzed conversion to pseudouridine, a post-transcriptional edit that upregulates mRNA translation. For this reason, T/U selection is not exchangeable: it is a deterministic binary switch that programs either catalytic DNA cleavage or translational RNA function in the same sequence skeleton.
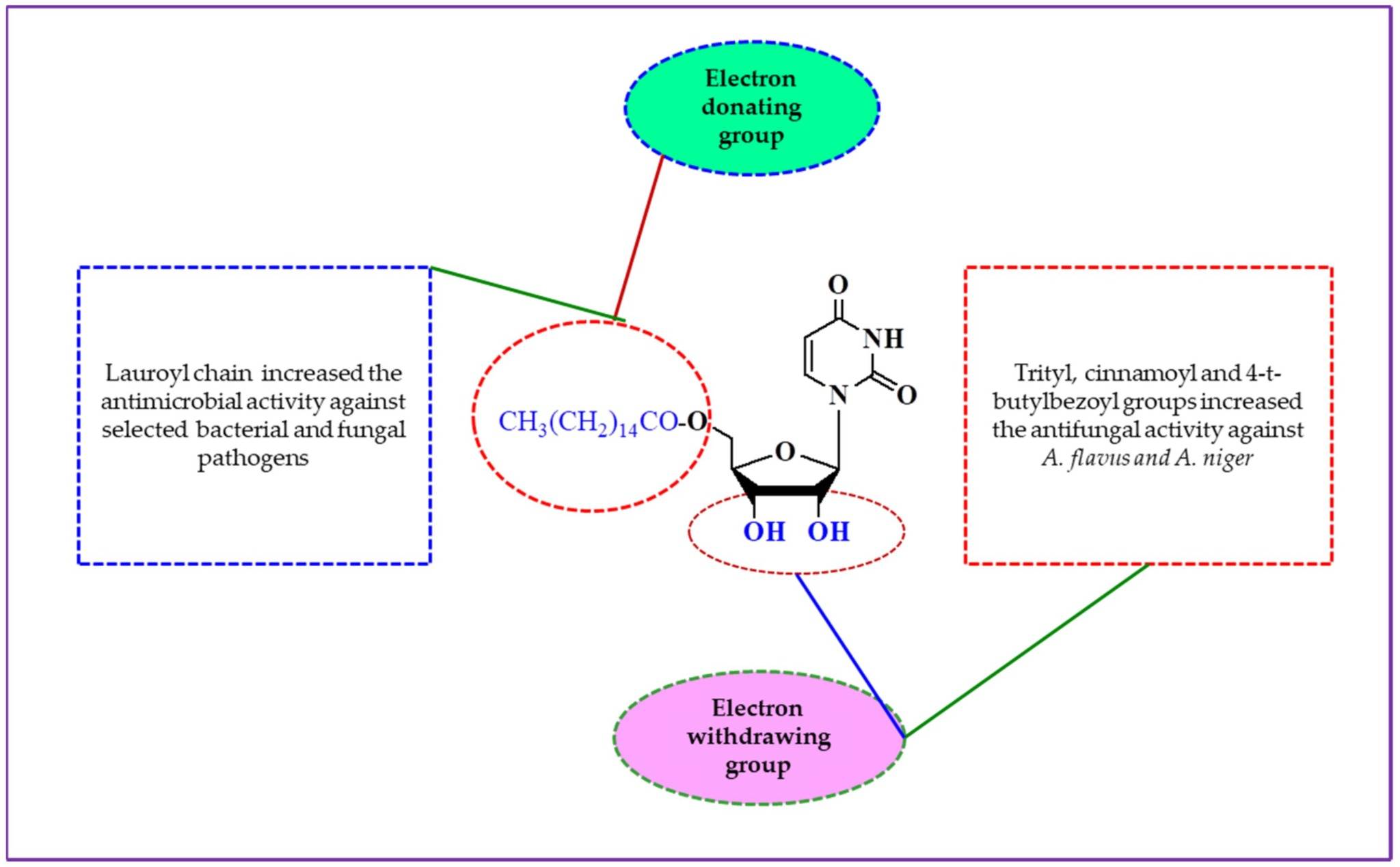 Fig. 1 Structure–activity relationship of the synthesized uridine derivatives.1,5
Fig. 1 Structure–activity relationship of the synthesized uridine derivatives.1,5
The presence of thymidine's 5-methyl adds hydrophobic surface area which allows crystallization from ethanol–water and reduces hygroscopicity for drum storage; the absence of the methyl in uridine renders it more water-soluble and hygroscopic, thus requiring inert-atmosphere packaging and cold-storage in warehouse. The 2'-OH group of uridine also contributes a superfluous hydrogen-bond donor which increases the boiling point of the protected intermediates and makes them more difficult to vacuum-dry, and thus often needs to be distilled at reduced pressure. During phosphoramidite synthesis, the presence of the 5-methyl in thymidine sterically hinders the N3 position and quells otherwise competing N-alkylation side reactions that afflict uridine amidites and thus contributes to a higher crude purity and less chromatography load. In this way the mere structural difference between T and U ripples beyond the biochemical into solvent choice, drying strategy and waste-disposal decisions throughout multi-kilogram scale campaigns.
For gapmer antisense platforms, thymidine is essential since the DNA-like region must attract RNase H while the 2'-MOE-uridine "wings" on either side provide higher affinity. Mutation of the thymidine to uridine in the central region prevents catalytic protein recruitment and results in loss of knock-down activity. For mRNA vaccines, uridine is completely substituted with pseudouridine or N1-methyl-pseudouridine to avoid PKR and RIG-I activation, modifications that are not possible on thymidine's methyl platform. Uridine is also required in siRNA duplexes for seed-region conformational entropy, but thymidine may be included at non-seed positions to increase melting temperature without increasing non-specific argonaute interactions. Circular RNA templates use uridine for ribozyme-mediated self-cleavage via the 2'-OH, a modification which is unavailable on thymidine. The choice of which sugar to use is driven by the platform's biology, not chemical ease.
Thymidine and uridine are chemically related pyrimidine nucleosides that are separated by a single methyl group and an oxygen atom. Thymidine contains 5-methyl-2'-deoxyribose, which mandates C2'-endo B-form geometry and recruitment of RNase-H, while uridine contains ribose with C3'-endo A-form pitch that is required for RISC and ribosome function. This small change is thus magnified into orthogonal biogenesis, stability, and regulatory mechanisms.
The two bases have a common six-membered diazine ring with keto-enol tautomerism; thymine has a 5-methyl addition group that enhances the π-stacking and reduces the hydration of the base, and uracil has an open C5-H that is useful for pseudouridine or 5-propynyl modifications. The exocyclic C4-carbonyl and N3-imine are the hydrogen-bonding anchors to adenine, but the 5-methyl in thymine sterically protects the N3 from indiscriminate alkylation in phosphoramidite formation, giving a higher purity crude. The polarity of the C2-carbonyl is also a determinant of metal-chelation propensity and can play a role in how well residual catalyst can be removed in downstream processing. Therefore the ring functional group pattern encodes both Watson–Crick interaction accuracy and selective synthetic handle affinities.
The 5-methyl increases van-der-Waals contacts that increase melting temperature and decrease breathing, but at the same time sterically occludes the C5-position for orthogonal post-synthetic click handles. In uridine, the vacant C5-H allows a palladium-catalyzed iodination that could be followed by Sonogashira lipidation to tune liver tropism without having to rely on auxiliary carriers. Metabolically, the methyl sterically hinders deamination to improve the plasma half-life, but makes the base more prone to oxidation into the 5-hydroxymethyl lesion which must be scavenged <0.1 % of the product to remove flags for genotoxicity. Crystallographically, the methyl forces the two adjacent base pairs to tilt by 10°, resulting in a tighter major groove with an improved protein readout. The methyl, therefore, is not just an inert placeholder, but a pharmacological rheostat for stability, conjugation and immunogenicity.
Ribose has a hydroxyl group at the 2'-carbon which fixes the sugar conformation in the north-domain (C3'-endo) which narrows the minor groove and coordinates with Mg2+, allowing 2'-O-methylation or 2'-fluorination, which protects siRNA from serum degradation. The absence of this 2'-oxygen in deoxyribose sugar allows for conformational flexibility in the south-domain (C2'-endo) that broadens the major groove, which forms a perfect platform for recognition by RNase-H. The ribose 2'-OH group also creates a nucleophilic attack site and is subject to a 2'-O-transesterification reaction during solid phase synthesis so is protected by fluoride-labile silyl ethers, which is not the case for deoxy intermediates, and this additional protection step in the former lengthens the synthesis cycle time. From a synthetic chemistry perspective, the absence of the 2'-OH in the deoxy sugar also makes the pKa of the adjacent 3'-OH 0.5 pKa units lower, which results in faster phosphoramidite coupling reaction under mildly basic conditions.
Thymidine nucleobases are the canonical pyrimidine backbones for DNA-like oligonucleotides. In addition to simply boosting the base-stacking energy and sequestering the deoxyribose from spontaneous deamination, the presence of the 5-methyl pre-conditions the inherent metabolic stability, RNase H recruitment and organic-solvent tolerance without impacting the amidite coupling cycle; this 1-atom edit provides a single building block that can simultaneously feed into solid-phase, antisense synthesis and enzymatic gapmer production, while imprinting both the chemical and manufacturing identities into the therapeutic scaffold pre-scale-up.
Thymidine is the default pyrimidine for DNA gapmers since its 5-methyl group enhances van-der-Waals contacts in the major groove to increase melting temperature while preserving RNase H competence; the same methyl group also inhibits spontaneous cytosine deamination, reducing mutation load during long-term storage. Because thymidine is conjugated to 2'-deoxyribose, it adopts C2'-endo pucker that both widens the major groove and creates the B-form geometry required for polymerase recognition, illustrating that base selection pre-defines both mechanism (cleavage vs translational) and chemistry route (organic vs aqueous) before the first coupling cycle.
5-methyl-thymidine is the native monomer, 5-fluoro-thymidine increases electron-withdrawal and strictness of base-pairing fidelity, and 4-thio-thymidine incorporates a photoswitchable chromophore that can be activated by near-IR light to release on-demand cleavage. The modifications are incorporated as phosphoramidites with standard protecting groups, so that the same resin provides both gene-silencing activity and optical control in one-pot workflow compatible with existing GMP lines.
Crystalline thymidine amidites are delivered under nitrogen with ≤0.2 % moisture and have ≥24 month ambient shelf lives. The 2'-hydroxyl's absence reduces base-catalyzed cleavage, which simplifies warehouse logistics and extends shelf life. The ability to store the same solid at room temperature, without the need for cold-chain infrastructure, also allows manufacturers to collapse inventory complexity into one ambient drum, which can be fed directly into the synthesizer without inline desalting or pH adjustment. This contrasts with the situation for ribonucleosides, whose hygroscopicity and counter-ion requirements make them much more complicated to handle.
In gapmer antisense oligonucleotides, native thymidine is often included in the center DNA core to preserve RNase H recruitment, while 5-methyl-thymidine is used in the wings to make the duplex more stable without hindering cleavage; for aptamers, 5-fluoro-thymidine can be photo-crosslinked to target proteins, permanently covalently locking it in the target binding pocket, and thus increase affinity. As these base edits are orthogonal to the phosphoramidite cycle, developers can toggle between catalytic cleavage or high-affinity recognition by simply changing the amidite bottle, providing a chemically deterministic means to program either knock-down or recognition within the same hardware platform.
Uridines represent the traditional pyrimidine base of RNA drugs. The ribose cis-diol locks RNA into C3'-endo A-form, the C5-H position is a modifiable handle to add immune silent modifications and the N3-imine tethers Watson–Crick basepairing; together these properties allow for siRNA cleavage, mRNA translation and aptamer folding all of which can be synthesized at scale by enzymatic polymerization.
Native uridine has a single-ring pyrimidine with C2=O and C4=O carbonyls and an N1-glycosidic nitrogen. These features impose a C3'-endo ribose pucker that is essential for ribosome decoding and RISC loading. The lack of a 5-methyl group reduces metabolic cost and enables wobble pairing with G or A to further increase codon degeneracy. Because it is not recognized by TLR7/8, uridine does not engage innate immune sensing, allowing high-dose RNA therapeutics without the need for adjuvant co-formulation. Finally, its UV absorbance at 260 nm is largely unaffected by most C5 edits, which allows for real-time UV monitoring during solid-phase synthesis.
Modified nucleosides such as pseudouridine (Ψ) rotate the glycosidic bond to C5–C1', which forms an additional hydrogen bond, increases Tm, and sterically shields the base from RIG-I, and is now routinely incorporated into clinical mRNA. 5-methyl-uridine (m5U) further attenuates TLR7 sensing without compromising translational efficiency. 5-fluoro-uridine increases duplex stability and RNase H cleavage specificity, which can be leveraged in the wings of gapmers. 2-thio-uridine enhances base-stacking and G-quadruplex stability, which can be exploited in the stems of aptamers. These edits are introduced during solid-phase phosphoramidite chemical synthesis, and can be incorporated one-pot, in lieu of natural nucleosides, into long (>100 nt) RNA transcripts without the need for post-synthetic chemical modifications, thus compressing cost and time-to-clinic.
5-methyl-uridine and pseudouridine (red) dampen TLR7/8 activation sterically, while 2-thio-uridine (blue) increases melting temperature without significant immunogenicity. Due to susceptibility of uridine to deamination after long exposure to acid conditions, high-purity specifications (<0.1 % pseudouridine isomers) are now written into release criteria to circumvent off-target innate sensing. 5-azido-uridine (purple) can be used to introduce click-chemistry handles for post-synthetic fluorescent or affinity tagging, to enable real-time tracking of deposition without altering biological function.
In small interfering RNA (siRNA), the presence of 2'-O-methyl-uridine in the guide strand seed region reduces off-target hybridisation, while 5-methyl-uridine in the passenger strand dampens Toll-like receptor 7 (TLR7) sensing, enabling subcutaneous dosing. In messenger RNA (mRNA), pseudouridine and N1-methyl-pseudouridine can be incorporated co-transcriptionally to improve translational efficiency and lower immunogenicity. In RNA aptamers, 5-fluoro-uridine can be used to enhance G-quadruplex stability, while 5-azido-uridine can be used for photo-cross-linking to enable target validation. Uridine's ability to adopt both C2'-endo and C3'-endo puckers (due to the fact that it is the only RNA base without an amino group at C2) is exploited by using it as a conformational "switch" which allows aptamers to fold into the complex tertiary structures necessary for high-affinity binding.
The decision between thymidine and uridine is not simply a "nitrogen-free base change": this chemical toggle predetermines synthetic route, duplex conformation and innate-sensor interaction (a 5-methyl group in thymidine induces C2'-endo pucker and organic-solvent miscibility required for solid-phase DNA assembly, while a 5-proton in uridine biases for C3'-endo pucker and aqueous solubility necessary for enzymatic RNA synthesis; being aware of this limit will enable developers to flow-crystallize thymidine amidites into RNase-stable gapmers and hold hygroscopic uridine triphosphates for immune-evasive mRNA, thus engraving both mechanism and manufacturability onto the same nucleoside core).
Table 2 Manufacturing dichotomy between T and U
| Parameter | Thymidine (T) | Uridine (U) | Downstream consequence |
| C-5 substituent | Methyl | Proton | DNA vs RNA fidelity |
| Sugar pucker | C2'-endo (south) | C3'-endo (north) | Duplex rigidity |
| Process solvent | Organic | Aqueous | Chemistry route |
| Innate sensor | Minimal TLR7/8 | RIG-I substrate | Immunogenicity dial |
Thymidine phosphoramidites enjoy the stabilizing effects of the 5-methyl group, which sterically hinders the N3, thereby reducing side reactions of N-alkylation during tetrazole activation and enhancing crude purity following oxidation. The 2'-H also prevents oxidative cleavage of cis-diols, and so if any periodate or Fenton reagents remain from sugar synthesis, these will not lead to chain truncation. Uridine amidites, by comparison, leave the C5-H vulnerable to accidental iodination if trace metal catalysts remain, leading to 5-iodo side-products that reduce coupling yields and fluoresce under UV-detection. The 2'-OH of uridine also allows the formation of intramolecular hydrogen bonds to the phosphate group, which transiently shields the reactive centre and retards the 3'-phosphitylation step; this is circumvented by increasing the concentration of tetrazole, but this comes at the cost of more acid waste and greater DMT-cation removal load. Thymidine cycles therefore tend to run faster and cleaner than their uridine counterparts, which require more rigorous metal scavenging and moisture control to achieve the same stepwise efficiencies.
5-Methyl makes it possible to avoid benzoyl protection of the N3-imine during solid-phase synthesis, since the base is less nucleophilic; only 5'-DMT and 3'-phosphoramidite require transient masking. Uridine requires two protective groups: benzoyl on N3 to avoid transamidation during deprotection of ammonia, and TBDMS or TOM on the 2'-OH to prevent acid-catalysed 2'-O-acylation. These additional manipulations add to reagent mass and solvent volume per kg of oligonucleotide. In addition, the benzoyl protecting group on uridine is base-labile but acid-stable, so when removing the DMT it is necessary to use the harsher trichloroacetic acid instead of the milder dichloroacetic acid used for thymidine. This is disadvantageous, because of the higher risk of depurination. This also causes a deviation in protecting-group choreography: thymidine enjoys a lean two-mask sequence, while uridine requires three-mask orthogonality, which complicates automation and lengthens cycle time.
Thymidine's 2'-H removes the cis-diol, allowing it to persist to lengthy aqueous work-ups and to prevent 2'-O-transesterisation that would otherwise lead to strand breakage under basic conditions. The 5-methyl group also reduces the hydration of the major groove and thus limits hydrolytic attack of the glycosidic bond; the depurination rates are cut in half compared to deoxyadenosine under the same exposure to acid. Uridine, in contrast, bears a 2'-OH that, under slightly alkaline pH and with the presence of trace Mg²⁺, forms a cyclic phosphate that causes strand scission and shorter full-length product. The unsubstituted C5 position is also susceptible to oxidative hydration, creating 5-hydroxy-uridine which tautomerises and impairs base-pairing. The ingress of moisture during storage in drums hydrolyses the cyanoethyl phosphate protecting group (unintentionally and prematurely) to the H-phosphonate diesters that will readily fragment during ion-pair HPLC. For these reasons, uridine derivatives require inert-atmosphere packaging, refrigerated storage and real-time water speciation, while thymidine intermediates are stable under ambient humidity for years.
Selecting thymidine or uridine is not an arbitrary design decision—it dictates duplex rigidity, intrinsic-sensor interaction and ribosome decoding capacity: 5-methyl in thymidine biases toward C2'-endo pucker and B-form helices that both recruit RNase H and protect DNA from cytosine deamination, while 5-proton in uridine biases toward C3'-endo pucker and north-conformation helices that are necessary for siRNA seed flexibility and for codon-anticodon wobble; appreciating this biophysical constraint enables the premeditated design of both pharmacodynamic activity and pharmacokinetic stealth prior to the first in animal dose.
Pseudouridine (Ψ) adds an additional imino proton and a C5–C1' glycosidic bond, which constrains the sugar into C3'-endo and increases helical twist. This has the effect of increasing melting temperature without increasing steric bulk. 5-Methoxy-uridine, on the other hand, rigidifies the base pair through π-conjugation, which decreases the entropic cost of target annealing and sharpens seed-region specificity in siRNA. Dihydrouridine (D), by contrast, saturates the C5=C6 double bond, inverting the sugar to C2'-endo and introducing local flexibility that is permissive for tertiary contacts in tRNA elbow regions; when engineered into aptamer stems, it speeds conformational switching required for protein recognition. These modifications are generally maintained despite preserving Watson–Crick hydrogen bonding and can be translated into slower off-rates, lower dosing frequency and better tissue persistence across RNA modalities.
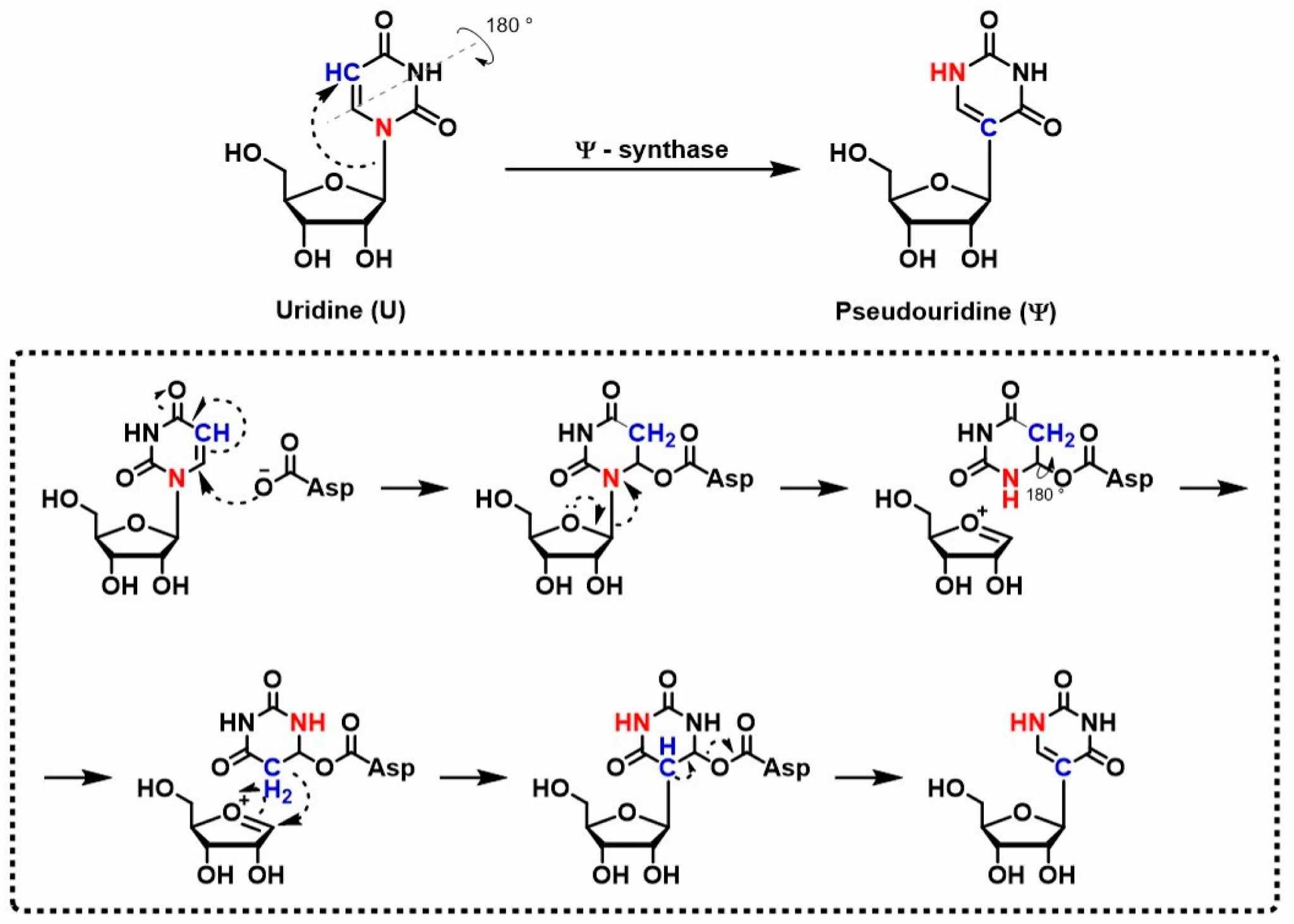 Fig. 2 Example of the mechanism of Ψ/U conversion catalyzed by PUS1.2,5
Fig. 2 Example of the mechanism of Ψ/U conversion catalyzed by PUS1.2,5
Ψ and N1-methyl-pseudouridine (m1Ψ) both enhance ribosome pausing density, thereby promoting re-initiation and producing 5- to 10-fold higher protein yields than native uridine, which has been leveraged in self-amplifying mRNA vaccines. 5-Propynyl-uridine deepens the major groove, and the concomitant increase in van-der-Waals contacts with argonaute residues speeds up RISC loading and precludes any off-target passenger-strand activity. In the context of circular RNA, m1Ψ impairs the activation of dsRNA sensors, resulting in a weaker innate immune response and less translational inhibition, which collectively improve protein expression both in vitro and in vivo. On the other hand, retention of native uridine in guide RNAs used for CRISPR ensures authentic Cas binding, whereas replacement with Ψ in the tracrRNA scaffold precludes interferon induction. These examples collectively show that uridine edits are pharmacological rheostats that tune gene-silencing potency, protein yield and immune compatibility within the same sequence backbone.
In contrast to DNA, RNA should not only consider the correct catalytic geometry but also immune stealth: Ψ and m1Ψ prevent TLR3/7 and RIG-I activation, but do not impair eIF4E binding, suggesting a possibility for repeated dosing without causing interferon tolerance. 2'-Fluoro-uridine can rigidify the seed to protect it from RNase A cleavage but preserve C3'-endo stacking that is not possible with thymidine. Dihydrouridine (D) imparts local flexibility that is crucial for tRNA elbow kissing, a tertiary interaction required for decoding by the ribosome; when applied to aptamer loops it speeds up conformational transitions needed for protein recognition. The C-glycosidic linkage of Ψ also prevents acid-catalysed depurination, a stability advantage during low-pH endosomal passage. Overall, these RNA-exclusive properties make modifications to uridine the most important knobs for tuning stability, specificity and immunological quietness of therapeutic RNAs.
Base selection is the irreversible fulcrum upon which hangs the decision to pursue an oligonucleotide as a DNA drug (driven by thymidine chemistry, recruit RNase-H, B-form) or as an RNA drug (driven by uridine chemistry, A-form, translatable); this choice multiplies into unique synthetic chemistries, impurity thresholds, stability margins and regulatory filing classes, making it the initial engineering decision that must be made well before toxicology or CMC timelines are defined.
In DNA, thymidine is preferred: its lack of a 2'-hydroxyl, and the 5-methyl group that it carries, both contribute to C2'-endo sugar pucker and preclude cytidine deaminase, respectively. Thymidine incorporation enables the use of gapmer antisense or gene-editing guides that are designed to induce transcript removal through RNase-H mediated catalytic cleavage. In RNA, on the other hand, the 2'-hydroxyl present on uridine (or the pseudouridine proxies used for it) induces C3'-endo conformation that is required for loading into RISC, decoding on ribosomes, and bypassing innate immune sensors to a large degree. Replacing uridine with thymidine in an mRNA backbone switches off translation and instead activates TLR3, while replacing uridine with thymidine in a gapmer backbone takes away the ability to recruit RNase-H and collapses its potency. Maximum lengths are different too: DNA ASOs can be 30mers with 2'-OMe flanks, while self-amplifying RNA vaccines are in the kilobase range and can only be synthesized via enzymatic polymerization of uridine monomers at a high rate. The DNA-vs-RNA decision is thus not a matter of style; it is a deterministic choice point that defines backbone composition, modification frequency, and target application.
Early-stage INDs can be established with research-grade amidites of ≥98 % purity, but from Phase II forward, chemists will need GMP-grade thymidine or uridine building blocks with ≤0.1 % free base, ≤1 ppm heavy metals and well-characterized anomeric purity of ≥99 % β-anomer to meet ICH Q11 drug-substance requirements. RNA drugs must also be able to explain to the FDA the intended rationale for each uridine modification (Ψ, m1Ψ, 5-OMe) in terms of TLR silence, translational enhancement and metabolite safety, whereas DNA drugs need to provide kinetic data on depurination and recruit data on RNase-H activation for thymidine-rich cores. Platform designation submissions will also need a risk-based assessment of base-edited versus native sequences, which encompasses off-target hybridization predictions and immunogenicity panels. In this way, the decision on the base is in the regulatory DNA of the programme long before the first-in-human dosing decision.
Costs of goods sold for thymidine pathways function as organic commodity chemicals: crystallization from ethanol–water, room-temperature storage, incinerable waste and incumbent supplier base ensure that COGS are low even at multi-ton scale. In contrast, uridine derivatives—especially Ψ or m1Ψ triphosphates—demand aqueous ion-exchange purification, freeze-drying, −20 °C cold-chain and RNAse-free suites, which appreciably raises variable cost and capital depreciation. Yield profiles are divergent as well: solid-phase thymidine gapmers incur linear loss on synthesis scale-up, whereas enzymatic uridine mRNA faces exponential drop-off beyond 4 kb, requiring downstream RNase III polishing and tangential-flow concentration. Scalability of the platform is thus tied to base selection: DNA indications can absorb higher cost-of-failure due to lower dosing, while RNA vaccines must achieve euro-per-dose pricing that only commoditized uridine feedstocks can provide. The T-vs-U decision, in other words, maps directly to facility design, environmental overhead and commercial viability.
We offer a comprehensive range of thymidine and uridine derivatives to support both DNA- and RNA-based therapeutic development. Our solutions are designed to help developers select the appropriate pyrimidine base for their platform while ensuring consistent synthesis performance, scalability, and regulatory readiness throughout the development lifecycle.
Our thymidine derivative portfolio is optimized for DNA-based oligonucleotide therapeutics, including antisense oligonucleotides and DNA aptamers. These materials are manufactured to high purity standards with strict control of impurities and moisture content, ensuring reliable performance during solid-phase synthesis. High-purity thymidine derivatives support efficient coupling, high sequence fidelity, and consistent batch performance, which are essential for both early-stage development and large-scale manufacturing. Native and protected thymidine forms are available to support phosphoramidite preparation and downstream synthesis workflows.
For RNA therapeutics, we supply a broad selection of uridine derivatives, including both native and modified forms tailored for siRNA, mRNA, and antisense RNA applications. Modified uridine derivatives are widely used to improve RNA stability, translational efficiency, and immunogenicity profiles. Our uridine derivatives are designed to be fully compatible with chemical synthesis and enzymatic processes, supporting flexible integration into different RNA production strategies. Controlled quality and consistent performance help ensure reproducible results across complex RNA drug programs.
Selecting between thymidine and uridine derivatives is a critical decision that impacts both drug performance and manufacturability. Our technical team provides expert guidance on base selection, taking into account therapeutic modality, synthesis strategy, modification requirements, and scale-up considerations. We support customers with monomer selection, protecting group compatibility, and process optimization, helping to align raw material choices with long-term development and regulatory goals. This collaborative approach reduces technical risk and supports efficient program progression.
We provide reliable and scalable supply of thymidine and uridine derivatives to support programs from preclinical research through GMP manufacturing. Our materials are supported by clear specifications, traceability, and appropriate documentation to facilitate smooth transitions between development stages. With a focus on long-term supply continuity and quality consistency, we help ensure that base selection decisions made early in development remain viable as programs advance toward clinical and commercial manufacturing.
If you need assistance selecting thymidine or uridine derivatives for your DNA or RNA therapeutic program, our experts are ready to help. Contact us today to request technical information, samples, or quotations, or to discuss optimized base selection strategies tailored to your development and manufacturing needs.
References
Thymidine is used in DNA, while uridine is used in RNA.
No, uridine is the correct pyrimidine base for RNA therapeutics.
Yes, modifications help reduce immunogenicity and improve stability.
Yes, it influences protecting group strategies and coupling performance.
The choice depends on whether your platform is DNA- or RNA-based.


 Loading ......
Loading ......