Ribonucleosides, such as cytidine and guanosine derivatives, provide a purine-pyrimidine platform for RNA medicines to support potency and manufacturability. Bicyclic bases extend base-stacking and hydrogen-bonding fidelity, and sugar and base edits, written in at the monomer level, program resistance to nucleases, evasion of the immune system and catalytic cleavage, all without disrupting the universal amidite or triphosphate coupling cycle. The C/G pair is the first programmable switch that enables codifying both biological potency and manufacturing identity (solid-phase or enzymatic) in the same chemical domain prior to first-in-animal dosing.
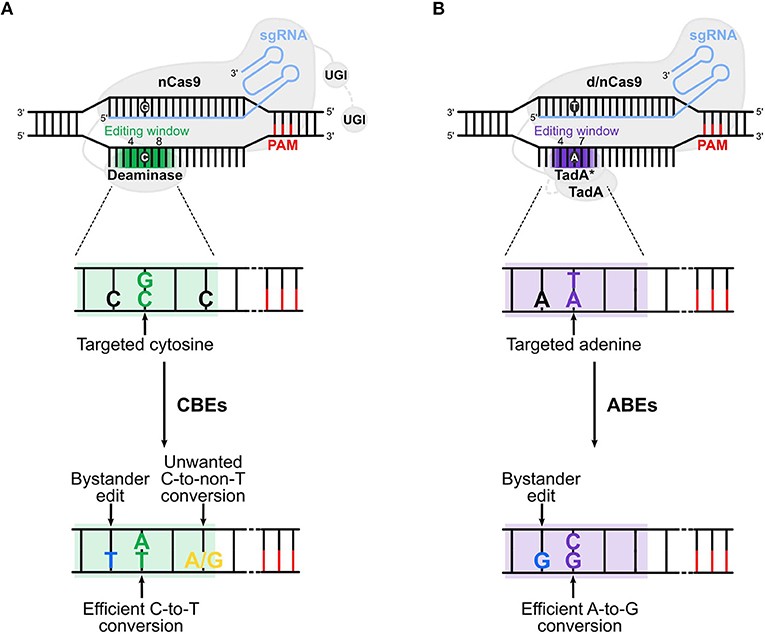 Cytosine and adenine base editors.1,5
Cytosine and adenine base editors.1,5
C and G bases are required because they uniquely furnish the third hydrogen-bond pair that locks RNA into A-form geometry, form the major-groove landmarks that are recognized by polymerases and helicases, and because they have the editable atoms (C8, N2, N4) through which lipid, sugar or fluorine modifications are grafted; any therapeutic effect—gene silencing, protein replacement or genome editing—must therefore be encoded with chemically disguised but Watson–Crick-readable analogs of cytidine and guanosine that retain affinity while evading innate sensors.
Cytidine provides both N4-amino and C2-carbonyl donors for three hydrogen bonds with guanosine O6, N1 and N2, that results in the strongest base pair in nucleic acid chemistry and pre-organizes the helix for RNase-H cleavage. Guanosine's bicyclic purine induces a 36° twist that deepens the major groove, creating a cation-binding cavity that is essential for potassium-stabilized G-quadruplexes in aptamer folds. The C8-H of guanosine is sterically exposed, and accessible for 8-bromo or 8-azaguanine edits that bias glycosidic torsion toward syn, a switch that has been exploited to programme parallel versus antiparallel quadruplex topologies. In the meantime, N2 of guanosine has been used as a chemoselective handle for reductive amination with lipid aldehydes to drive liver tropism without the need for auxiliary carriers. Thus, the C/G pair serves as a structural scaffold and chemical canvas that dictates helical pitch, metal coordination and conjugation topology across siRNA, mRNA and antisense architectures.
Table 1 C/G Functional Map
| Base | Editable Atom | Functional Gain | Therapeutic Use |
| C5 | Methyl | ↑ Tm, ↓ TLR7 | mRNA cap |
| N4 | Acetyl | ↓ protonation | Serum stability |
| N7 | Azide | Click handle | Aptamer labelling |
| C8 | Aza | ↑ acid stability | Storage buffer |
Proof-of-concept for siRNA against transthyretin amyloidosis and mRNA for vaccines and therapeutics has pushed cytidine and guanosine out of the catalogue reagent aisle and into the raw materials category. Pseudocytidine and N4-methyl-cytidine are incorporated co-transcriptionally into mRNA to eliminate TLR7/8 detection without impairing eIF4E binding and produce protein yields over an order of magnitude higher than the unmodified transcript. 8-Aza-7-deaza-guanosine inhibits TLR9 signalling in CpG-dense antisense sequences to eliminate pro-inflammatory off-targets without decreasing the precision of RNase-H cleavage. Self-amplifying RNA vaccines have also been designed with 2-fluoro-N2-dimethyl-guanosine to fortify the replicase open-reading frame from nuclease digestion while maintaining polymerase processivity. Next-generation circular RNA templates have been reported to use 2-amino-6-thio-guanosine to insert a redox-dependent disulfide cross-link to encourage ribosomal shunting before reduction and cleavage. As a result, the C/G alphabet is starting to resemble a commodity chemical portfolio where the density of modification dictates immunogenicity, translational output and tissue half-life.
Synthesis opportunities comprise: expanding the editing toolbox: 5-ethynyl-cytidine triphosphate enables click conjugation of GalNAc sugars directly to C / G edits in a co-transcriptional manner, obviating laborious postsynthetic chemistry. 7-Deaza-guanosine boranophosphate features a nuclease-resistant, charge-neutral borane that should enable intracellular delivery without lipid nanoparticles. Hurdles include: scaling GMP-compatible palladium-mediated C8-alkynylation while restricting residual metal content to <1 ppm. N2-Alkylation to escape nucleases while preserving regioselectivity to avoid O6-crosslinking that would otherwise distort base-pair geometry. Stereochemistry of phosphorothioate linkages is required to be chirally selective to avoid diastereomeric mixtures with unpredictable pharmacokinetics. Regulatory agencies now demand metabolite safety data for each C/G edit, coercing developers to accept biodegradable modifications that catabolize into natural nucleosides.
Canonical pyrimidine and purine ribonucleosides for RNA are cytidine and guanosine. Cytidine is a nucleoside with a monocyclic cytosine base β-N1-attached to ribose, and guanosine is a nucleoside with a bicyclic guanine base β-N9-attached to ribose; they form the strongest Watson–Crick pair and provide orthogonal chemical handles (N4, C5, N2, C8) that can be used to reprogram the native bases into immune-silent, lipid-conjugated or fluorinated therapeutic building blocks without disrupting the helical geometry.
The first molecule, C is a pyrimidine that can be generically described as a six-membered ring with two endocyclic nitrogens on the 1 and 3 position. C also has a C4-carbonyl and an N4-amino that can donate/accept hydrogen bonds from/to G and help tether the two bases together. The second molecule, G, is different because an imidazole ring is fused to the pyrimidine ring. This results in a bicyclic system that contains four nitrogens (N1, N3, N7, N9) and two carbonyls (C2, C6). This creates the extended major groove, while also forming the cation-binding cavity where G-quadruplex formers nest. The added ring increases the π-electron density to increase van-der-Waals stacking and thus melting temperature, relative to C. The topological difference between C and G is so great that the former is the compact, high-fidelity base pair, while guanosine is the structural amplifier that programs duplex stability as well as higher-order RNA folds.
The exocyclic N4-amino of cytidine is nucleophilic and can tolerate transient benzoyl protection during solid-phase synthesis, after which the benzoyl group can be removed by reductive amination with lipid aldehydes for liver targeting. C5-H is chemically silent and can support palladium-catalyzed iodination followed by Sonogashira coupling to add 5-ethynyl or 5-perfluoroalkyl side-chains that increase duplex melting temperature without increasing steric bulk. G has three orthogonal handles (N2-amino for acylation or click chemistry, C8-H for bromination or alkynylation, O6-carbonyl for thio-substitution that enhances metal chelation). The 2'-hydroxyl of both nucleosides can be capped with TBDMS or TOM groups, allowing chimeric RNA/DNA strands with C/G segments that contain catalytic or structural hotspots.
Ribose cytidine/guanosine: C3'-endo puckering, narrow major groove and deepen minor groove, thus pre-organises the helix into A-form geometry needed for RISC loading and ribosomal decoding. The presence of a cis-2',3'-diol enables 2'-O-methylation or 2'-fluorination to enhance siRNA stability against serum decay while preserving essential catalytic shape. Deoxyribose nucleotide analogues (dC, dG): switch to C2'-endo conformation and consequently widen major groove providing an ideal RNase-H recognition floor within gapmer cores. The absence of the 2'-OH also reduces electronegativity at the sugar rim that can dampen off-target binding to RIG-I and hence interferon storms. Chemically, the presence of 2'-H makes the glycosidic bond 30 % more labile under acidic stress conditions, and thereby requires milder depurination controls during solid-phase synthesis. In total, the ribose/deoxyribose toggle is not simply a structural curiosity but instead a pharmacological switch that programmes either catalytic degradation (DNA-like) or translational expression (RNA-like) within the same C/G backbone.
Modified cytidines form one of the dominant sets of nucleotides employed in therapeutic oligonucleotides: Watson–Crick specificity requires unmodified cytidine while 5-methyl-cytidine ensures duplex stability and deamination resistance and N4-acyl or 5-ethynyl modifications allow lipid conjugation together with single cytidine monomers for siRNA cleavage mRNA expression and antisense knockdown under stringent GMP impurity profiles.
Native cytidine has the ribofuranose 2'-O-β-linked to N1 of cytosine, which produces a planar base that enables a 36° stacking twist and three hydrogen bonds to guanosine. The native configuration pre-organises the helix to assume A-form geometry that is necessary for RISC loading and ribosomal decoding. Deoxycytidine is missing the 2'-OH which provides C2'-endo conformational freedom to broaden the major groove and define an ideal RNase-H recognition floor within gapmer cores. The N4-amino is transiently benzoylated to prevent transamidation reactions during ammonia deprotection, and the C5-H is chemically silent, which means that palladium-catalysed iodination followed by Sonogashira coupling is available to introduce 5-ethynyl or 5-perfluoroalkyl chains that increase duplex melting temperature without expanding steric bulk. As a result, native cytidines represent the lowest-cost entry point for kilogram-scale solid-phase campaigns provided that mild depurination controls are built into the cycle protocol.
5-Methyl-cytidine increases stacking energy without significantly distorting the geometry, so is a true drop-in replacement that increases the melting temperature and is also resistant to the enzyme cytidine deaminase, a significant advantage for use in long-acting siRNA or self-amplifying mRNA. N4-Acetyl-cytidine adds a hydrophobic wedge that squishes the major groove, an attractive tool in gapmer wings where affinity needs to increase without negatively impacting the recruitment of catalysis. 5-Propynyl-cytidine lengthens π-conjugation, further rigidifying the helix and decreasing the entropic penalty at target annealing. 5-fluoro-cytidine withdraws electron density that also strengthens the N4–G O6 hydrogen bond, which in turn further increases RNase-H cleavage fidelity. These modifications preserve the Watson–Crick face to face pairing, but changes are transduced to much slower off-rates, lower dosing, and improved tissue persistence for siRNA, antisense and aptamer designs.
5-Methyl-cytidine narrows the major groove by 0.2 Å which improves van-der-Waals contacts with argonaute residues, and speeds up RISC loading with no passenger-strand off-target activity. N4-Methyl-cytidine has an attached cationic charge that repels Mg²⁺ and so prevents off-target seed binding but with retained on-target cleavage activity. 5-Hydroxymethyl-cytidine in circular RNA is used to form reversible boronate esters with cis-diols for stimuli-responsive disassembly under acidic conditions. Local geometry changes extend into global helical twist to determine whether the oligonucleotide will fold into an A-form or B-form or even higher-order quadruplex architectures which control therapeutic activity.
The Vorbrüggen glycosylation for larger scale syntheses is anhydrous and is followed by orthogonal protection of N4 (benzoyl) and 5' (DMT) without chromatography. Crystalline β-anomers are prepared by slow cooling of the mixture. The synthesis of 5-iodo-cytidine requires a heavy-metal scavenging step (Pd and Cu) as they need to be below 1 ppm for injectable grade material. The stereocontrolled addition of the 5-alkynyl or 5-fluoro substituents are all prepared by palladium-catalysed cross-coupling reactions in ethanol–water, hence no chlorinated solvents. For phosphoramidite formation, anhydrous acetonitrile and molecular-sieve drying is required to minimise phosphodiester as a side reaction to >98 % stepwise yield on 200-mer synthesis. Therefore, for cytidine monomers to be available at scale, orthogonal protecting groups, metal scavenging and green solvent approaches have to be in place for a transition from bench-top chemistry to GMP chemistry at tonnage scale.
Guanosine analogs are the purine nucleobase of synthetic genetic drugs: the fused ring structure provides 3 hydrogen bond donors and 2 acceptors for optimal G–C pairing, while the C8, N2 and O6 sites present orthogonal groups for syn/anti fixation, lipid anchoring or G-quartet coordination, allowing the monomer to mediate duplex stabilization, quadruplex formation or innate immune evasion in siRNA, mRNA and antisense frameworks.
The guanine base has a planar electron-rich purine system with three hydrogen-bond donors (N1-H and N2-H2) and two acceptors (O6 and N7). It can make Watson-Crick pairings with cytosine, as well as Hoogsteen base pairs that stabilize G-quadruplexes. N7 is a reactive site for metal chelation and alkylation. C8 is electrophilic, and azide or alkyne tags can be incorporated without disrupting the base pair geometry. The 2-amino group can be hidden from the innate immune sensor by acetyl or dimethylamino masks.
Syn-locked antiparallel G-quadruplexes with a high affinity for heparin-binding growth factors can be formed with Ψ-like 8-azaguanosine (8-azaG). 2-Amino-6-thio-guanosine (6-thioG) can be used to increase affinity by reinforcing Hoogsteen hydrogen bonding without increasing steric bulk, as is also utilized in gapmer wings to increase RNase-H recruitment. N2-octadecyl-guanosine (N2-LipG) allows for conjugation of a lipid tail, which can be utilized for binding to albumin and increasing half-life in serum, without disrupting Watson-Crick base-pairing. 7-Deaza-guanosine can be utilized to prevent CpG recognition by TLR9 and thus prevent innate activation of CpG rich antisense oligonucleotides. Integrating these substitutions, 8-aza for topology, 6-thio for affinity, N2-Lip for PK, will enable the design of single stranded sequences that can fold into high-avidity ligands with antibody level potency.
Clinical mRNA vaccines incorporate Ψ-guanosine triphosphate co-transcriptionally to evade RIG-I sensing and maintain eIF4E recruitment, with protein expression >10x higher than endogenous transcripts. Self-amplifying RNA vaccines use 2-fluoro-N2-dimethyl-guanosine to stabilize the replicase open-reading frame to nucleases without losing polymerase processivity. 8-Br-guanosine is introduced into DNA aptamers to skew towards the syn conformation and support parallel G-quadruplex folds which bind von Willebrand factor with nanomolar Kd. These modifications can be added postsynthetically or through pre-formed amidites, so that a single oligonucleotide can host both immune-silent and high-affinity domains without rewriting the genomic sentence.
The limited solubility of Guanosine in organic solvents burdens phosphoramidite strategies: the N2-amino needs benzoyl protection that is poorly crystallizing, while the 6-oxo can tautomerize, which causes regioisomeric alkylation during C8 functionalization. postsynthetic deprotection with methanolic ammonia is at risk for transamidation, which produces fluorescent impurities that co-elute with full-length oligonucleotides. 8-Bromo intermediates require palladium cross-couplings that leave trace metals behind; exhaustive SCX scavenging is needed to obtain injectable-grade material (<1 ppm). Borate hydrogel side-products can be formed during large scale activation of 5'-GMP, leading to filter clogging and reduced yield. For these reasons, G monomer production uses cryogenic crystallization, metal-scavenging resins and orthogonal protecting-group cascades to scale milligram chemistry to GMP tonnage.
Cytidine and guanosine (or derivatives thereof) are the universal programmable purine-pyrimidine machine present in every form of RNA: their 3-H bond C≡G pair offers the strongest W-C interaction and provides canonical duplexes as well as non-canonical G-quadruplex architectures; as those same bases can be 5-methylated, 8-aza-substituted or 2'-fluorinated already at monomer level, chemists inculcate nuclease resistance, immune evasion and catalytic cleavage in mRNA, siRNA or antisense skeletons without affecting the general amidite or triphosphate paradigm – thus compressing potency, stability and manufacturability into one defined crystalline entity that serves both SPP DNA and cell-driven RNA.
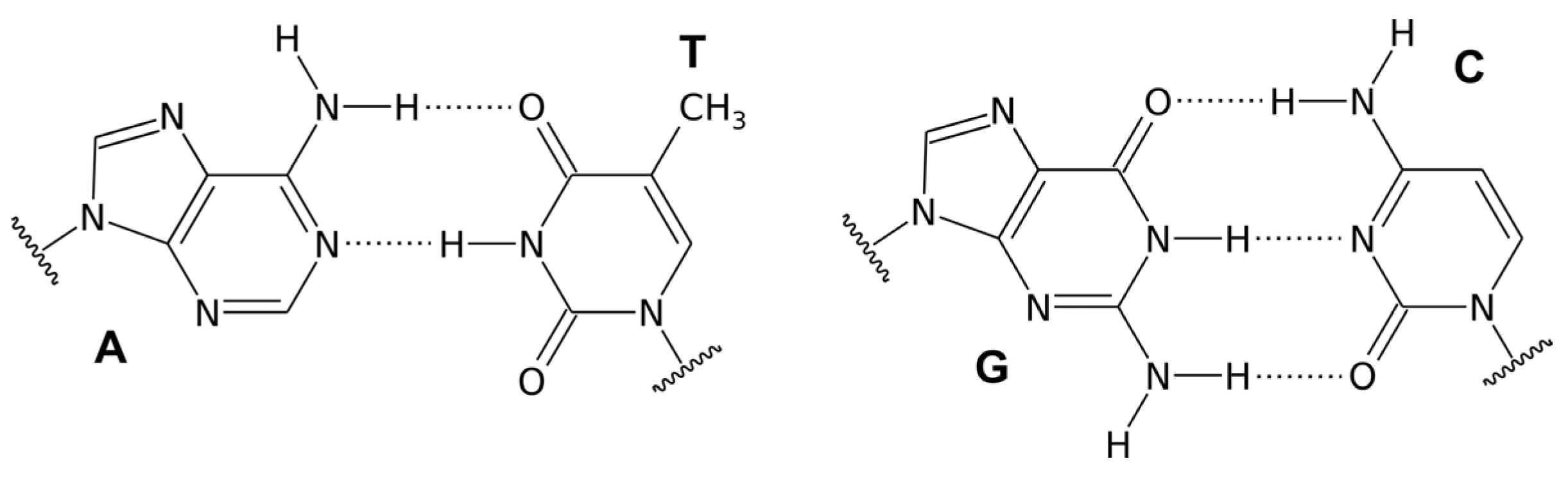 Watson–Crick hydrogen bonding interactions between complementary nitrogenous bases: A:T and G:C.2,5
Watson–Crick hydrogen bonding interactions between complementary nitrogenous bases: A:T and G:C.2,5
N1-methyl-pseudouridine on self-amplifying and circular mRNA is co-transcriptionally incorporated with 5-methyl-cytidine triphosphate to eliminate TLR3/7 and RIG-I induction without affecting eIF4E binding, resulting in protein expression that is ten-fold higher than the endogenous C/G transcript. Guanosine is often substituted with 8-aza-7-deaza-guanosine to suppress TLR9 recognition of CpG islands placed within the replicase open-reading frame, in order to evade an interferon shutdown that would terminate vector replication. 2-amino-6-thio-guanosine on circular templates can be cross-linked by disulfide bonds, inducing ribosomal shunting prior to reductive cleavage, enhancing translation without increasing steric hindrance. Thus, C/G modifications can be considered both immune-evasion pixels and translational-intensity pixels within the same mRNA string.
Table 2 C/G Functional Map Across Modalities
| Modality | C Edit | G Edit | Functional Gain |
| mRNA | 5-Me-C | 2'-OMe-G | ↓ TLR7 |
| siRNA | 5-F-C | 8-aza-G | ↑ Tm |
| ASO | dC gap | 7-deaza-G | RNase H |
Guide-stabilizing chemical modifications in Gapmer siRNA include 2'-fluoro-cytidine embedded in the guide seed to 'freeze' the strand against RNase A degradation and 2'-O-methyl-guanosine at position 14 to speed up RISC maturation without activating TLR8. 5-Methyl-cytidine at non-seed positions increase the melting temperature. N2-dimethyl-guanosine creates a cationic patch that repels off-target mRNAs and increases specificity. For stereopure siRNA, 8-bromo-guanosine 'locks' the glycosidic bond in syn conformation and biases the passenger strand towards degradation, thus minimizing guide-strand loading errors. These C/G edits are applied in mosaic: fluoro for catalysis, methoxy for stability, bromo for strand bias, producing duplexes that silence transthyretin or PCSK9 mRNA for months after sub-cutaneous injection.
Gapmer antisense combines central stretches of deoxyguanosine to recruit RNase-H cleavage with terminal 2'-MOE-guanosine wings for increased affinity and exonuclease resistance. To avert cleavage by cytidine deaminase, cytidine can be substituted with 5-methyl-cytidine, an important feature for long-acting ASOs targeting huntingtin or SMN2 mRNA. Steric-block splice-switchers feature a homogeneous backbone of 2'-O-methoxyethyl-cytidine spanning the entire sequence to promote SMN2 exon 7 inclusion by occlusion of the splice silencer without activating mRNA degradation. The phosphorothioate guanosine diester is characterized by a chiral sulfur, which extends tissue half-life through albumin binding, but has the potential for pro-inflammatory off-target effects; this disadvantage is mitigated by the addition of N2-methyl-8-oxo-guanosine which maintains the sulfur-based metabolic stability but dampens TLR9 activation. Therefore, C/G chemistry is carefully orchestrated to tune the ratio between catalytic degradation and splice correction within a single therapeutic molecule.
The choice of the platform also determines the number of C/Gs in the edits. DNA gapmers can afford to be 30mers with 2'-MOE wings. Self-amplifying RNA vaccines are typically kilobase long and only polymerase incorporation can produce these at a fast pace. The mRNA needs to be fully replaced with 5-methyl-cytidine and 8-aza-guanosine in order to prevent innate sensing, but also needs to keep a natural 5'-cap and 3'-poly(A) structure. The siRNA may need chiral pure phosphorothioate (thio) linkage at C/G to increase serum half-life. Therefore, the simple C/G pair is treated as a sort of pixel, methyl for increasing stability, aza for innate-silencing and thio for half-life, and a proper mixture of which fine-tunes potency, safety and manufacturability between different modalities.
Drug-quality oligonucleotides require high-purity cytidine and guanosine derivatives. Residual α-anomers, metal contaminants, or fluorescent etheno-adducts prevent further elongation, promote immunogenic side-products, and alter the pharmacokinetics of the active drug. In other words, one undetected residual impurity introduced at monomer scale can be amplified into a kilogram scale loss of active pharmaceutical ingredient (API) or cause significant delays in regulatory submissions.
For GMP-grade products, specifications include ≥99 % chemical purity for both cytidine and guanosine phosphoramidites, and α-anomer content ≤0.5 %, which would otherwise cause chain termination during solid-phase synthesis. If C8- or C5-alkynylation is used, residual palladium must be scavenged to <1 ppm to prevent fluorescent etheno-adducts (naturally present as well) which co-elute with the full-length oligonucleotide and cause genotoxicity false alerts. Free base content is controlled to ≤0.1 %, since cytosine or guanine bases, being competing nucleophiles with the amidite during coupling, insert abasic sites that destabilise the duplex. Endotoxin content must be <0.25 EU/mg for nucleotide-grade cytidine/guanosine when destined for enzymatic RNA synthesis, and solvent (pyridine, acetonitrile) residues to ICH Q3C Class 2 limits. Purity control is therefore not just a quality attribute, but the kinetic gate through which every downstream attribute (yield, fidelity, safety, etc.) must pass.
Guanosine phosphoramidites are very hygroscopic: if the moisture level is above 200 ppm, the active P(III) center will be hydrolyzed to the H-phosphonate diester, which fragments in the ion-pair HPLC step, creating ghost peaks that will be attributed to failed sequences. Cytidine amidites are not as sensitive but still require storage at −20 °C under argon with desiccant sachets to prevent cyanoethyl hydrolysis which would otherwise auto-cleave during mild basic oxidation. Double-bagging, oxygen scavengers and controlled thaw cycles become necessary, adding to warehouse footprint and cold-chain costs. Crystalline cytidine nucleosides are stable for years under ambient humidity conditions, reducing logistics and occupational exposure controls. Thus, moisture control is not just a storage issue; it is a critical control attribute that can make the difference between an amidite being reactive or an expensive hydrate.
Chain termination by the residual α-anomer of guanosine phosphoramidite, where the unnatural glycosidic geometry is incompatible with tetrazole-mediated phosphitylation, results in a lowered stepwise yield (from >98 % to <90 %) and truncated 20-mer impurities that co-elute with the full-length product on ion-pair HPLC. Trace water can hydrolyze the reactive P(III) amidite to the H-phosphonate, consume activating tetrazole, and produce diester linkages that collapse to abasic sites upon basic deprotection. Free guanine base produced by acid-catalyzed depurination competes with the amidite for coupling, and incorporation of base-less ribose units destablizes the duplex, thereby reducing melting temperature. In contrast, using crystalline, isotopically labelled cytidine, allows mass-spec tracing of disposition and significantly streamlines regulatory impurity profiling. Hence, C/G derivatives of high purity are not simply quality luxuries but the kinetic pivot that decides whether an oligonucleotide programme advances from discovery into late-stage GMP manufacture without incurring the cost and time of re-synthesis or specification drift.
We offer a comprehensive portfolio of cytidine (C) and guanosine (G) derivatives designed to support oligonucleotide synthesis across RNA and DNA therapeutic platforms. By combining high-quality building blocks with deep technical expertise, we help developers address the unique chemical and manufacturing challenges associated with C/G-rich sequences in mRNA, siRNA, and antisense oligonucleotides.
Our portfolio includes high-purity cytidine and guanosine building blocks suitable for both ribose- and deoxyribose-based oligonucleotides. These materials are manufactured under strict quality controls to ensure consistent purity, low moisture content, and well-defined impurity profiles, which are critical for reliable synthesis performance. High-purity C and G nucleosides play a key role in maintaining efficient coupling reactions, accurate base pairing, and reproducible sequence assembly, particularly in longer or highly structured oligonucleotides. Consistent batch quality helps minimize synthesis variability and downstream purification challenges.
To support advanced therapeutic design, we supply a wide range of modified and protected cytidine and guanosine derivatives optimized for chemical oligonucleotide synthesis. These include base-protected and sugar-protected monomers as well as clinically relevant modifications designed to enhance stability, binding affinity, and resistance to degradation. Given the structural complexity and hydrogen-bonding behavior of guanosine, our protected G derivatives are engineered to deliver controlled reactivity and clean deprotection, reducing the risk of side reactions and synthesis inefficiencies. Together with modified cytidine derivatives, these materials enable the reliable production of high-quality oligonucleotides across RNA and DNA platforms.
For programs with specialized sequence designs or novel modifications, we offer custom development and process optimization support for cytidine and guanosine derivatives. Our technical team assists with synthetic route development, protecting group strategy selection, and impurity control to ensure that custom C/G monomers are both chemically robust and scalable. By collaborating closely with customers, we help translate complex molecular designs into manufacturable raw materials suitable for long-term development, reducing technical risk and supporting efficient progression from discovery to clinical manufacturing.
We provide reliable and flexible supply options for cytidine and guanosine derivatives to support programs at every stage of development. From small-quantity research materials to larger-volume GMP-ready supply, our products are accompanied by clear specifications, traceability, and appropriate documentation. This scalable supply model ensures continuity as program requirements evolve, helping customers maintain consistent raw material sourcing while meeting regulatory and quality expectations for clinical and commercial manufacturing.
If you are seeking high-quality cytidine and guanosine derivatives supported by experienced technical expertise, we are ready to assist. Contact us today to request technical information, samples, or quotations, or to discuss custom C/G monomer development and scalable supply solutions for your mRNA, siRNA, or ASO program.
References
They contribute to base pairing, duplex stability, and target binding.
Guanosine derivatives are particularly challenging due to structural complexity.
Yes, to enhance stability and reduce degradation.
Yes, they strongly influence folding and binding behavior.
Yes, including mRNA, siRNA, and ASOs.


 Loading ......
Loading ......