Chemical modification of oligonucleotides changes their pharmacologic properties: it stabilizes the phosphate backbone, replaces immunostimulatory nucleobases with inert analogs, and pre-structures the ribose conformations that are needed for high-affinity binding to the target. Modifications enable a long serum half-life, high sequence selectivity, and repeated systemic administration without immune activation–properties that have helped transform oligonucleotides from an experimental class of drug to a new generation of medicines.
Base modifications are necessary as unmodified A, U, C and G nucleotides are transient, immunogenic and thermodynamically unstable; chemically unmasked they activate TLR3/7/9, attract RNase A cleavage and fall out of register with their complementary strands, making base editing the first—and irreversible—design decision that determines whether an siRNA, mRNA or antisense molecule will circulate, enter cells and engage its target safely.
Native (naked) nucleic acids are quickly sensed, excreted, and cause an immune response when delivered systemically. The 2'-OH ribose rings in unmodified RNAs mediate 2'-O-transesterification by serum ribonucleases (RNAses) leading to 3'-phosphate containing fragments that are renally excreted within minutes. Pathogen-associated molecular patterns such as the exocyclic N3-H and N1-H of uridine and guanosine are recognized by endosomal TLR7/8 leading to IFN-α, IL-6 and TNF-α pathways that both inactivate the drug and create flu-like side effects. B-form breathing allows off-target seed pairing and non-physiological gene silencing or translation repression. Anionic phosphates result in albumin repulsion and increased glomerular filtration, increasing the necessary dose. These problems with naked nucleic acids prevent their use with repeat systemic dosing and forced chemists to shield every nucleophilic atom while maintaining Watson–Crick readability.
It started with first-generation phosphorothioate backbones that substituted non-bridging oxygens with sulfur to increase serum half-life but trigger inflammatory off-targets. Second-generation 2'-O-methyl and 2'-fluoro sugars then followed to further rigidify the backbone without disrupting catalytic geometry. This third generation is based on replacing heteroatoms within the base itself: pseudouridine and N1-methyl-pseudouridine substitutes N1-H with C–C or N–Me bonds to remove TLR contact while simultaneously tightening C3'-endo stacking; 5-methyl-cytidine and 8-aza-guanosine increase melting temperature and prevent deamination. Locked and unlocked sugars (LNA, UNA) also tune the desired rigidity allowing for gapmer cores to recruit RNase H while wings are nuclease-proof. These combinatorial edits are now co-transcriptionally installed, which leads to long mRNA or self-amplifying RNA that can express protein for days while still avoiding innate sensors.
Table 1 Generation of Modified Bases
| Generation | Key edit | Liability solved | New issue |
| 1st | Phosphorothioate | Nuclease resistance | Inflammation |
| 2st | 2'-OMe, 2'-F | TLR silence | ↓flexibility |
| 3st | Ψ, m5C, 8-aza | Immune invisibility | Synthetic complexity |
Clinical translation has been demonstrated using siRNA targeting transthyretin, with 2'-OMe and 2'-F edited cytidine/guanosine. This modification enabled a 12-month knock-down with a single infusion, demonstrating base editing as a practical long-term therapeutic strategy. mRNA vaccines for COVID-19 were developed by swapping out uridine for N1-methyl-pseudouridine across the spike transcript, which completely removes IFN induction while retaining expression of the target antigen. This allowed billion-dose production with no red flags for reactogenicity. Gapmer ASOs are also utilizing 5-methyl-cytidine and 8-aza-guanosine in order to silence huntingtin or SMN2 transcripts over the course of months, minimizing the need for frequent dosing and hepatotoxicity. Base-edited aptamers that target von Willebrand factor are able to bind their target with nanomolar affinity, and do not require backbone phosphorothioation, suggesting that heterocycle edits themselves are capable of creating antibody-like potency. These therapeutic milestones have helped re-establish base editing from an academic novelty to a core component of nucleic acid therapeutics.
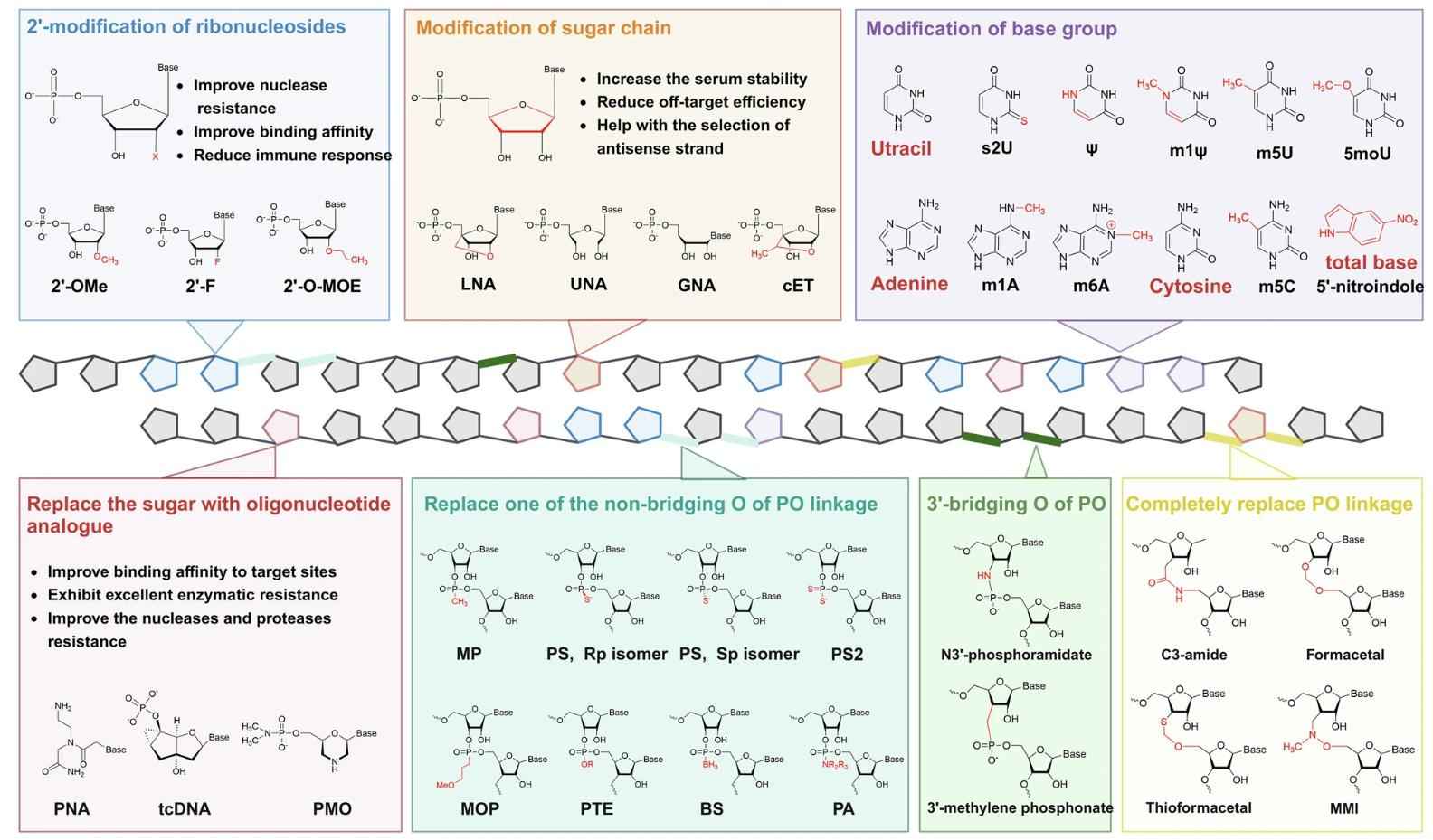 Fig. 1 Chemical modifications used for small nucleic acids.1,5
Fig. 1 Chemical modifications used for small nucleic acids.1,5
Chemical alterations of the nucleic acid pharmacopeptide: They stabilize labile phosphate backbones by nuclease resistance; they replace immune activating heterocycles with silent analogs; they pre-orient sugar puckers for high affinity target binding. The resulting increase in serum half-life, sequence specificity and lack of inflammation, following repeated systemic administration, have resulted in the development of the first oligonucleotide therapeutics.
Bases that are already present as naturally occurring modifications (pseudouridine, 5-methyl-cytidine, N6-methyl-adenosine) in cell RNA can be fully substituted as they will be recognised as "self" by the TLR or RIG-I systems. Synthetic nucleotide bases such as 8-aza-guanosine, 5-propynyl-uridine or 7-deaza-adenosine are not expected to cross-recognize endogenous nucleic acids or proteins but contain heteroatoms or alkyne handles that are not found in nature, providing higher Tm or functional click handles. Safety data on the metabolites will be required for the regulators. The line is a blurry one: pseudouridine is natural but must be chemically installed, while 5-ethynyl-uridine is unnatural but is metabolized to uracil. For this reason, developers have to provide data on the biotransformation pathways of each base edit, regardless of whether it is nature inspired or completely synthetic, in order to support chronic dosing regimens.
By replacing uridine with pseudouridine (Ψ), one abolishes TLR7/8 activation while tightening C3'-endo stacking. This edit is now the backbone of all mRNA vaccines. 5-Methyl-cytidine and N4-methyl-cytidine both confer resistance to cytidine deaminase and increase melting temperature without increasing steric bulk and are workhorses of gapmer antisense. N6-Methyl-adenosine also preserves eIF4E recruitment but silences RIG-I allowing self-amplifying mRNA to express antigen for days without an interferon flare. 8-Aza-7-deaza-guanosine evades TLR9 recognition of CpG islands thereby removing pro-inflammatory off-targets while also retaining RNase-H cleavage fidelity. All of these edits can be installed co-transcriptionally (via the triphosphate analogue) or postsynthetically via click chemistry allowing a single oligonucleotide to bear both immune silent and high affinity segments without having to rewrite the genomic sentence.
Base edits are the "invisible ink" – the ribosome/pol reads the code but the innate sensors see the sequence as self. This approach has the potential to provide the best therapeutic index with the least steric penalty. Sugar edits (2'-F, 2'-OMe, LNA) rigidify the backbone but limit flexibility and can result in off-target argonaute contacts if used too liberally. Backbone thioation can prolong half-life but can run the risk of pro-inflammatory sulfur chemistry and requires chiral control. As a result the current approach layer base edits for immune silence with sugar edits for nuclease resistance and minimal backbone thioation for renal retention to create synergistic potency without additive toxicity. This triumvirate of base for silence, sugar for stability and backbone for PK has become the default starting point for 3rd generation Oligo therapeutics.
Base edits are a form of molecular camo: they sterically protect against nucleases, pre-set sugar puckers for tighter stacking and remove labile protons prone to hydrolysis, thus conferring a dramatic increase in serum half-life from minutes to days without increasing overall size or compromising Watson–Crick readability.
Chemical modification at the 2' position: Substitution of 2'-hydroxyl with 2'-O-methyl or 2'-fluoro enforces C3'-endo pucker to mask the ribose from ss-endonucleases without disrupting the A-form geometry necessary for loading onto RISC. Chemical modification at the N-position: Pseudouridine (ψ) and N1-methyl-pseudouridine lack the N1-H hydrogen-bond donor, thereby eliminating TLR7/8 binding, and reduce enzymatic docking. Chemical modification at the sugar: 5-methyl-cytidine and 5-propynyl-uridine extend π-conjugation, giving a hydrophobic shroud that sterically occludes the active sites of exonucleases. Chemical modification at the phosphate backbone: Phosphorothioate diesters insert a chiral sulfur atom that poisons 3'-exonuclease cleavage by altering electrostatics and binding kinetics. The combinatorial effects of these edits are cooperative: sugar hardening truncates the first cut, base lipophilicity prevents enzyme docking, and backbone thioation overcomes endonucleolytic nicks, producing oligonucleotides with days-long serum half-lives rather than minutes.
Examples include 5-Methyl-cytidine (5MeC) and 2-amino-adenosine. 5MeC increases melting temperature through better base-stacking properties and a dehydration of the major groove, while 2-amino-adenosine can form a third hydrogen bond with thymidine (also see 2'-hydroxyl-sugar bond in Locked nucleic acid). Addition of a propynyl group to the C5 position of cytidine or uridine, can conjugate the base pair further, to the point that base pairing is "pre-locked" and a smaller entropic penalty is paid when the two strands anneal. Locked nucleic acids (LNAs) "freeze" the sugar in the C3'-endo conformation, which shortens the phosphate backbone and increases the helical twist, with a resultant 3–5 °C gain per modified nucleotide. These modifications all preserve Watson–Crick geometry, but in aggregate can translate to measurable decreases in off-rates, which manifest as slower dosing frequencies and improved tissue persistence for siRNA, antisense and aptamer designs.
The analog 5-methyl-cytidine is a substrate-determinant mimic that is not recognized by cytidine deaminase because the enzyme's active site is sterically blocked. Cytidine deaminase is a proofreading enzyme that converts cytidine to uridine, which is a likely cause of sequence infidelity. The nucleoside pseudouridine has the N1-C1' glycosidic bond replaced with the more stable C5-C1' bond. This change is the removal of the acid-labile imino group, so nucleic acids containing pseudouridine are not subject to acid-catalysed depurination, which is a common fate of nucleic acids under low-pH endosomal conditions. The nucleoside 8-aza-7-deaza-guanosine has the basic N7 removed, making the molecule less vulnerable to oxidative depurination and it retains the ability to form the same hydrogen bonds as guanosine. Each of these modifications reduces the rate of spontaneous hydrolysis of the oligonucleotide by at least an order of magnitude, which increases the stability of the therapeutic oligonucleotide to its storage, freeze-thaw and shelf life conditions, as well as during long circulation times.
Base edits work like tunable affinity knobs: they pre-fold the sugar puckers for tighter stacking, remove labile protons that loosen hydrogen bonds, and add hydrophobic or π-extended groups that enhance van-der-Waals contacts, all of which synergize to increase melting temperature, sharpen seed specificity and enable single-digit nanomolar binding without increasing oligonucleotide length or losing sequence fidelity.
Pseudouridine (Ψ) has an additional imino proton and a C5–C1' glycosidic bond that constrains the C3'-endo stacking interaction. These structural changes increase the melting temperature while maintaining Watson–Crick geometry. 5-Methyl-cytidine constricts the major groove of RNA, resulting in increased π–π overlap with guanosine and a smaller entropic penalty upon annealing. Propynyl groups on C5 of cytidine or uridine increase conjugation and pre-orient the base pair leading to faster association kinetics. Such modifications result in a slower off-rate, reduced dosing frequency and increased tissue persistence in siRNA, antisense and aptamer designs.
The modified nucleotide N2-dimethyl-guanosine is a particularly useful modification to increase the specificity of siRNA. This modification creates a cationic patch that will sterically clash with any adenosine that is not Watson–Crick paired in the seed region, thus increasing seed-region specificity. The non-natural nucleotide 8-aza-7-deaza-guanosine, which lacks the basic N7 nitrogen, has been shown to specifically remove Hoogsteen pairing with off-target cytosine bases while maintaining Watson–Crick pairing fidelity. 5-Fluoro-cytidine, a modified cytosine nucleotide, is able to subtract electron density from the C–N3 site, thus fortifying the N4–G O6 hydrogen bond and eliminating G-U wobble. Such modifications result in an overall decrease in the permissiveness of seed mismatches, which results in decreased off-target effects and higher therapeutic indices without the need to redesign a given siRNA sequence.
Locked nucleic acids (LNA) at C/G positions lock the sugar in C3'-endo conformation. As a result, the helix is pre-bent and internal loops are exposed for protein recognition. This principle has been used for aptamers that bind to von Willebrand factor. 2-Amino-6-thio-guanosine incorporates a thiol which forms reversible disulfide cross-links and can kinetically trap a G-quadruplex fold. The structure unfolds in the reductive cytosol, which precisely releases the antisense strand inside the cell. Dihydrouridine at the C5 position of cytidine saturates the double bond, increasing local flexibility and enabling ribosomal scanning over structured 5'-UTRs in circular mRNA. Base edits are conformational switches that programme either rigidity or flexibility on demand to dictate whether the oligonucleotide is locked into a duplex or quadruplex, or remain as single-strand to be accessible to its cognate target.
Base editing "camouflages" viral RNAs by disrupting PAMPs: uridine to pseudouridine or N1-methyl-pseudouridine removes the N3-H hydrogen-bond donor that TLR7/8 and RIG-I recognise, 5-methyl-cytidine and 8-aza-guanosine eliminate CpG motifs sensed by TLR9, to avert type-I interferon induction and cytokine storms that normally eliminate the drug product and cause flu-like reactogenicity.
Table 2 Base-edit stealth map and mechanistic origin
| Base edit | Sensor target | Molecular mask | Clinical read-out |
| pseudouridine | RIG-I | C-C glycosidic bond | ↓ IFN-α |
| 5-methyl-C | TLR7/8 | Methyl steric shield | ↓ TNF-α |
| 7-deaza-G | TLR7 | Removed N7 donor | ↓ IL-6 |
| N6-methyl-A | PKR | Reduced eIF2α-P | ↑ protein output |
Endogenous RNA is sensed by endosomal TLR3/7/8 and cytosolic RIG-I/MDA5; unmodified uridine-rich stretches are potent agonists of these receptors, inducing IFN-α, IL-6 and TNF-α that both attenuate efficacy and cause systemic inflammation. The substitution of uridine with pseudouridine (Ψ) or N1-methyl-pseudouridine (m1Ψ) removes the N3-H donor required for TLR7 binding, while maintaining the Watson–Crick geometry required for ribosome decoding. 5-Methyl-cytidine and N4-acetyl-cytidine mask the major-groove edge recognized by TLR8, and 8-aza-7-deaza-guanosine disrupts the CpG motif essential for TLR9 activation. These edits do not abolish immune sensing completely (repeat dosing can still elicit low-level cytokines), but they shift the balance from inflammatory to tolerogenic, allowing chronic treatment without interferon fatigue.
Beyond innate receptor silencing, base edits directly improve physical tolerability. The C-glycosidic bond of pseudouridine prevents acid-catalysed depurination, accounting for lower injection-site rash observed with unmodified mRNA. 5-Methyl-cytidine decreases the isoelectric point of the transcript, resulting in less non-specific protein adsorption and complement activation. N1-Methyl-pseudouridine, by increasing ribosome pausing density, biases re-initiation and increases the protein output at a given dose, dampening dose-dependent side effects such as fever or myalgia. Delivery-vehicle reactogenicity can be indirectly reduced: lower mRNA doses require less lipid nanoparticle, limiting exposure to PEGylated moieties that can elicit anti-PEG antibodies and accelerated blood clearance. In summary, base edits function as systemic tolerability amplifiers and not simply innate immune silencers.
Over-modification has the potential to swing the pendulum to the other extreme: complete substitution of guanosine by 8-aza-G eliminates the G-quadruplex motifs that drive aptamer folding, and a high density of 5-methyl-cytidine flattens the major groove and hinders RNase-H binding to the gapmer core. Today, regulatory agencies require a risk-based table comparing each base edit with the pharmacodynamic endpoint of the edit (knock-down, translation, splicing) and its safety profile towards the metabolites that will arise from this edit. A de facto standard is emerging around "chemical parsimony" – use only the minimum number of base edits required to obtain immune silence and the desired potency, keep sugar modifications for nuclease resistance only, and restrict backbone thioation to PK tuning. This judiciously layered approach helps ensure that potency does not come at the cost of unanticipated toxicities, which helps maintain both the activity and tolerability of these agents with chronic dosing regimens.
Platform adjustments are actually modality-specific 'tuners': siRNA base mods strengthen the seed and 'mute' TLR7, ASO base mods cinch gapmer wings while leaving RNase-H cores intact, mRNA base mods swap uridine for immunologically 'invisible' analogues, and aptamer base mods pre-fold G-quadruplexes—enabling each platform to realize nanomolar potency without increasing dose or compromising safety.
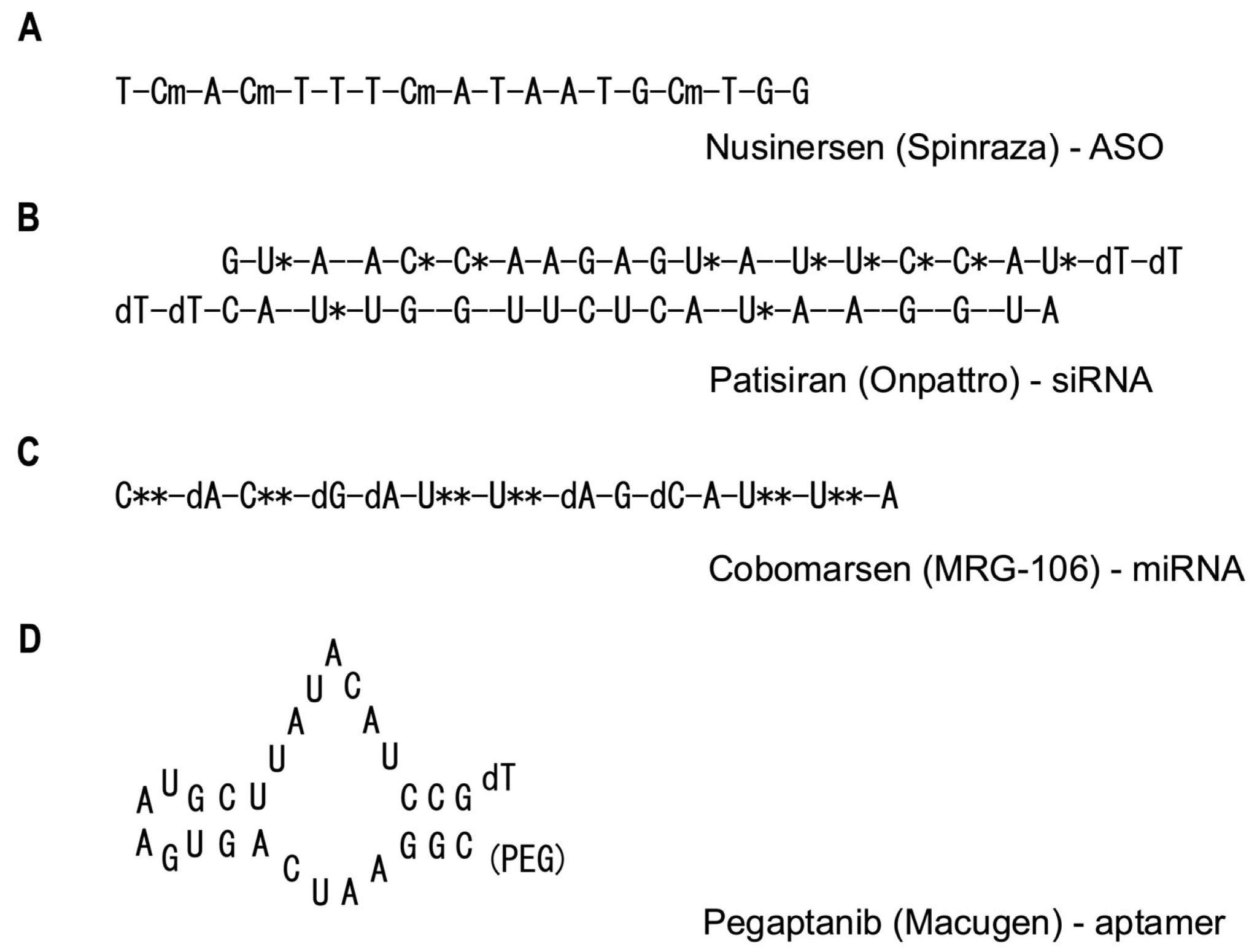 Fig. 2 Structural representations of representative API molecules for ASO, siRNA, miRNA, and aptamer-based therapeutics.2,5
Fig. 2 Structural representations of representative API molecules for ASO, siRNA, miRNA, and aptamer-based therapeutics.2,5
The various design layers of siRNA base edits are arrayed in a mosaic pattern. 2'-fluoro-uridine and 2'-O-methyl-cytidine are used to "harden" the guide strand with RNase A resistance, while still permitting A-form geometry and loading into RISC; N1-methyl-pseudouridine at non-seed positions abolishes TLR7/8 recognition without compromising catalytic turnover. 5-Methyl-cytidine at position 14 increases melting temperature to diminish off-target seed binding; 8-aza-7-deaza-guanosine interferes with Hoogsteen pairing with mismatched cytosine to enhance specificity. These are spread apart in a step-wise manner every third base to prevent excessive rigidity of the duplex. As a result, such duplexes can silence transthyretin or PCSK9 mRNA for months after sub-cutaneous injection, without causing interferon flare.
Gapmer ASOs contain 5-methyl-cytidine in their 2'-MOE wings, increasing affinity and blocking cytidine deaminase, while the internal tracts of deoxyguanosine recruit RNase H1 to cut the huntingtin or SMN2 transcript. 8-Aza-7-deaza-guanosine is placed in CpG islands to prevent TLR9 recognition, eliminating pro-inflammatory off-targets without reducing catalytic cleavage. Steric-block splice-switchers feature a homogeneous N4-acetyl-cytidine along the full length of the molecule, redirecting SMN2 exon 7 inclusion by blocking the splice silencer without eliciting mRNA degradation. These base modifications are combined with a small degree of backbone thioation just sufficient to delay renal clearance, creating 18-mer oligonucleotides that result in durable knock-down at sub-milligram levels.
Complete substitution of uridine for N1-methyl-pseudouridine and cytidine for 5-methyl-cytidine, eliminate TLR3/7/8 and RIG-I sensing, but retains eIF4E recruitment allowing self-amplifying mRNA to drive monoclonal antibodies to microgram-per-millilitre titres in serum. 8-Aza-7-deaza-guanosine breaks CpG motifs in replicase open-reading frame which would otherwise trigger interferon shutdown that collapses vector replication. For circular RNA, 2-amino-6-thio-guanosine allows for reversible disulfide cross-links that facilitate ribosomal shunting before reductive cleavage, increases translational yield without increasing steric bulk. The base edits are made co-transcriptionally by triphosphate analogues, which can result in long transcripts that evade innate sensors and have high translational efficiency.
An aptamer against the von Willebrand factor uses 8-bromo-guanosine to direct syn geometry, which stabilizes a parallel G-quadruplex fold with nanomolar Kd and does not require backbone phosphorothioation. Guide RNAs for CRISPR (circULAR single-stranded RNA guide) use Ψ-guanosine to disrupt TLR9 binding but maintain native Cas protein binding. RNA tiles for self-assembly use 5-propynyl-cytidine to sharpen loop angles and form hexagonal arrays that can be used to deliver siRNA aggregates without liposomes. These new classes of modalities are using base edits like design pixels: bromo to shape topology, thio to drive redox cleavage, propynyl to induce lattice rigidity, enabling novel therapeutic shapes that extend beyond traditional duplex or quadruplex architectures.
The synthesis of modified bases requires a departure from traditional nucleoside chemistry: each atom replacement (C8-aza, N1-methyl, 5-propynyl) necessitates additional protection–deprotection steps, metal-catalyzed couplings and regio-selective crystallizations which increase solvent mass and analytical load, transforming a three-step process into a ten-step enterprise where yield, metal-scrap and stereochemistry need to be nailed down well before kilo-scale GMP release.
Chemical synthesis of Ψ or m1Ψ starts from uridine and proceeds via C–N bond cleavage, tautomerisation and stereoselective C–C ribosylation catalyzed by Mn²⁺. Each additional methyl or propynyl group requires palladium cross-coupling at C5 followed by ion-exchange polishing to maintain residual Pd < 1 ppm. 8-Aza-7-deaza-guanosine needs regioselective bromination at C8 followed by Sonogashira lipidation in aqueous ethanol to prevent racemisation at the anomeric centre. Protecting-group orthogonality had to be extended from two (DMT, benzoyl) to four (TBDMS, trityl, alloc, acetyl) to survive the iterative acid, base and fluoride conditions. As a result, route scouting needs to optimize step-count with green-chemistry scorecards, enabling crystallisation of the final amidite rather than chromatography.
Typical specifications for GMP-grade modified bases include ≥99 % chemical purity, ≤0.5 % α-anomer and ≤1 ppm residual Pd, while Ψ triphosphates also require endotoxin <0.25 EU/mg and bioburden ≤10 CFU/g since they are introduced into enzymatic IVT without further purification. Analytical packages range from HPLC-UV to ion-pair UPLC-CAD for quantitation of regioisomers, ICP-MS for metal-trace analysis and ¹H-¹³C HMBC for verification of the C–C glycosidic linkage stereochemistry. Reference standards should be qualified against in-house primary lots, with clearly defined expiry and retest dates. A failure in the anomeric ratio or elemental impurity profile necessitates an investigation which can take weeks, so on-line NIR or Raman probes are appealing for tracking of tautomerisation and metal scavenging endpoints.
Modified bases are not commodity chemicals, so new synthetic routes must be qualified from the perspective of ICH Q11 starting-material guidances (kinase source, ATP regeneration chemistry, pyrophosphate fate-mapping, etc. for enzymatic IVT routes). Cold-chain logistics are tighter: 5-methyl-cytidine is stable at ambient humidity but 7-deaza-guanosine triphosphate must be stored at −20 °C and contained in glass-lined vessels to avoid metal leaching. The earlier the base identity is locked in with the intended modality (DNA vs RNA), the tighter the cost and tech-transfer timeline and the less likely the supply shock of a late-stage route switch.
We provide deep expertise in the development and supply of base-modified nucleoside building blocks to support high-performance nucleic acid drug development. By combining advanced chemistry capabilities with application-driven technical support, we help customers design and manufacture oligonucleotide therapeutics with improved stability, potency, and safety profiles.
Our portfolio includes a broad range of base-modified nucleoside derivatives covering all canonical nucleobases used in RNA and DNA therapeutics. These modifications are designed to address key limitations of native nucleic acids, including susceptibility to degradation, limited binding affinity, and undesired immune activation. The portfolio includes synthesis-ready nucleosides with clinically relevant base modifications, manufactured to high purity standards and compatible with solid-phase oligonucleotide synthesis. Consistent quality and well-defined specifications help ensure predictable performance during phosphoramidite preparation and oligonucleotide assembly.
For programs requiring novel or highly specific modifications, we offer custom nucleoside synthesis and base-modification strategy development. Our technical team supports the design and synthesis of tailored nucleoside derivatives, including modification placement, protecting group selection, and route optimization. By focusing on both chemical feasibility and manufacturability, we help ensure that custom base-modified nucleosides can be scaled reliably while maintaining performance and quality. This approach enables efficient translation from early molecular concepts to development-ready raw materials.
Base modification decisions have far-reaching effects on both drug performance and manufacturing efficiency. Our experts provide technical guidance throughout drug design and process development, including support for modification selection, synthesis compatibility, and impurity control. We work closely with customers to optimize coupling efficiency, deprotection conditions, and overall synthesis robustness, helping to reduce development risk and improve process consistency. This collaborative support ensures that base-modified nucleosides perform reliably under real-world manufacturing conditions.
We support nucleic acid drug programs across the full development lifecycle, from early discovery and lead optimization through clinical and GMP manufacturing. Our flexible supply model and technical expertise enable smooth transitions between development stages while maintaining consistent raw material quality. Materials are provided with appropriate documentation, traceability, and quality controls to support regulatory expectations and long-term manufacturing requirements. This end-to-end support helps ensure continuity and confidence as programs advance toward commercialization.
If you are developing nucleic acid drugs that require advanced base modifications, our team is ready to support your program. Contact us today to request technical information, samples, or quotations, or to discuss custom base-modified nucleoside solutions and scalable manufacturing support for high-performance RNA and DNA therapeutics.
References
Native nucleic acids are unstable and prone to degradation.
Yes, they improve binding affinity and target specificity.
Yes, certain modifications reduce innate immune activation.
Most clinically successful oligonucleotide drugs use modified bases.
They increase synthesis complexity but improve final drug quality.


 Loading ......
Loading ......