The standard alphabet of nucleic-acid therapeutics consists of the nucleoside derivatives of adenine, thymine/uracil, cytosine and guanine. Their sugar, base and phosphate chemistries can be atom-by-atom edited to create siRNA, antisense, mRNA and aptamer drugs that preserve genetic information, resist nucleases, avoid off-target hybridization and allow tissue-specific delivery—an engineering toolkit now propelling explosive growth in RNA-based medicine.
The four canonical nucleosides matter because they alone bridge biology’s information layer with medicinal chemistry’s manufacturing layer: their heterocycles encode sequence specificity, their sugars dictate helical geometry and metabolic half-life, and their phosphates provide attachment points for lipids, GalNAc or morpholino tails that steer biodistribution; any therapeutic outcome—knock-down, splice-switching or protein replacement—must therefore be written with chemically disguised yet Watson–Crick-readable versions of A, T/U, C or G.
Table 1 Canonical nucleoside derivatives and their therapeutic functions
| Base | Common modification | Therapeutic purpose | Analytical focus |
| A | N6-methyl, 2'-F | Enhances duplex stability | UV-melting curve |
| U/T | 5-methyl, 2-thio | Reduces innate sensing | LC-MS/MS ratio |
| C | 5-methyl, LNA | Improves nuclease resistance | RNase H assay |
| G | 8-aza, 2'-OMe | Increases base-pair fidelity | NMR imino region |
Therapeutic oligonucleotides are comprised of a four-letter alphabet whose sense is not lost even if each letter is decorated with 2'-fluoro, 2'-O-methyl, phosphorothioate or base modifications. Adenosine analogues can be N6-methylated to silence TLR7 signalling while preserving argonaute recognition; uridine can be swapped for pseudouridine or 5-methoxy-uridine to stiffen base-pair geometry and increase translational output; cytidine can bear 5 methyl or 5-hydroxymethyl groups that are deamination resistant while remaining susceptible to RNase H cleavage; and guanosine can be 7-deazated or 8-brominated to disarm innate sensors or stabilize G-quadruplex folds. Since all substitutions utilize the same hydrogen-bond grammar to be read, the therapeutic index can be finely adjusted without rewriting the genomic sequence while transforming the monomer set into an endlessly customizable linguistic toolkit.
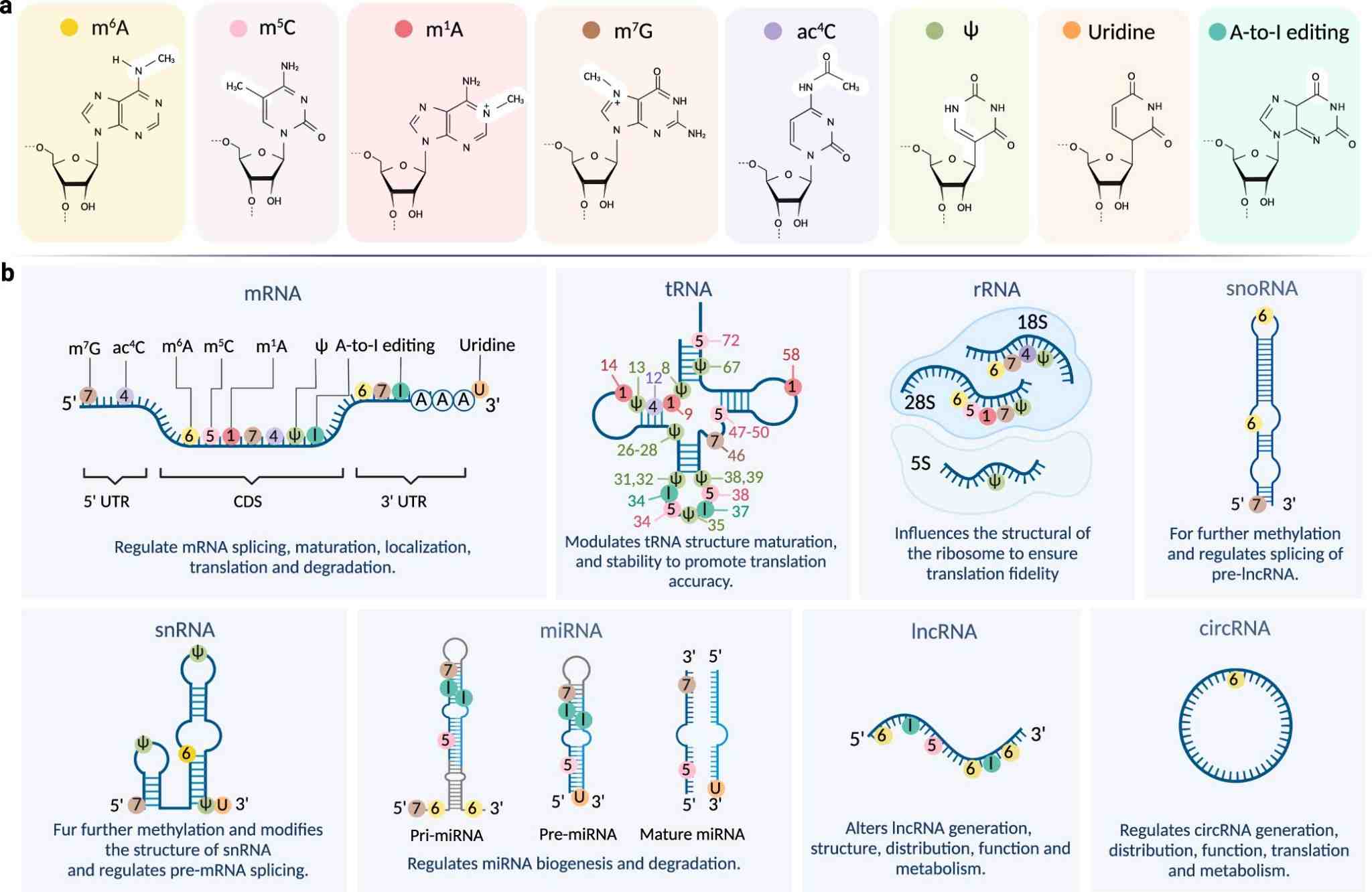 Fig. 1 RNA modifications and their distributions on different RNA subtypes.1,5
Fig. 1 RNA modifications and their distributions on different RNA subtypes.1,5
Genetic information is chemically encoded by reversing the canonical biosynthetic pathway, polymerizing phosphoramidites that are already masked with acid-labile dimethoxytrityl ethers, base-labile acyl protectors, and fluoride-labile silyl groups on the 2'-hydroxyl rather than triphosphates in cells. The synthetic grammar therefore consists of a nested protection scheme—acid for 5'-OH, base for exocyclic amines, fluoride for 2'-OH—that enables >99 % stepwise yield on the scale of a 200-mer mRNA. Post-synthetic deprotection, anion-exchange purification, and sterile filtration convert information into kilogram quantities of drug substance with molecular weight, stereochemistry and endotoxin load specified in pharmacopeial monographs, thereby compressing the digital genome into a shelf-stable, injectable chemical object for encapsulation in lipid nanoparticles.
The clinical development of siRNA against hereditary transthyretin amyloidosis, mRNA for preventive vaccines and antisense oligonucleotides for spinal muscular atrophy are turning catalogue reagents into critical raw materials. GMP-grade monomers with controlled residual solvents, trace metals and microbial burden are becoming regulatory prerequisites that are driving suppliers towards continuous-flow amidite synthesis, on-line analytics and single-use bioreactors. Self-amplifying RNA, circular RNA and bispecific aptamers will further diversify demand with their requirements for 5'-triphosphate analogues, 2'-amino-ribonucleosides and lipid-conjugated bases that are currently academic curiosities. The four-letter nucleoside alphabet is being turned into a commodity chemical portfolio whose capacity planning, supply-chain resilience and environmental sustainability determines how quickly genomic medicines can be distributed to global populations.
Canonical nucleosides – adenosine, guanosine, cytidine, thymidine and uridine – are the chemical alphabet of nucleic-acid therapeutics; their ribose or deoxyribose scaffolds can be locked, fluorinated, methylated or conjugated without altering the solid-phase phosphoramidite cycle, so the same automated synthesizer can be used to deliver siRNA, antisense, mRNA or aptamer drugs whose stability, immunogenicity and target affinity are pre-encoded at the monomer level – a plug-and-play modularity that collapses lead-time across multiple RNA platforms while maintaining a single, regulator-accepted manufacturing backbone.
The distinct structures of the two heterocyclic rings that comprise nucleosides allow for the selective harnessing of each in medicinal applications. Purine nucleosides (adenosine and guanosine) contain a fused imidazole-pyrimidine bicyclic structure, which confers greater π-surface area for stacking and van-der-Waals contacts in RNA helices, but also decreased water solubility and greater steric footprint; pyrimidine nucleosides (cytidine, thymidine, and uridine), with their single six-membered ring, are less able to form these interactions, but exhibit higher water solubility and a smaller steric footprint. These distinctions have been used to either adjust duplex rigidity, or to engineer gapmer wings with greater melting temperature. The purine scaffold provides the N6-alkyl or C-8-aza positions for modification that can enhance base-pairing fidelity without altering sugar pucker. The C-5 and C-2 positions on the pyrimidine can accommodate a thio, methyl or propynyl group that can increase hydrophobic burial and decrease off-target hybridization. These modifications provide orthogonal handles to improve potency in each of these classes.
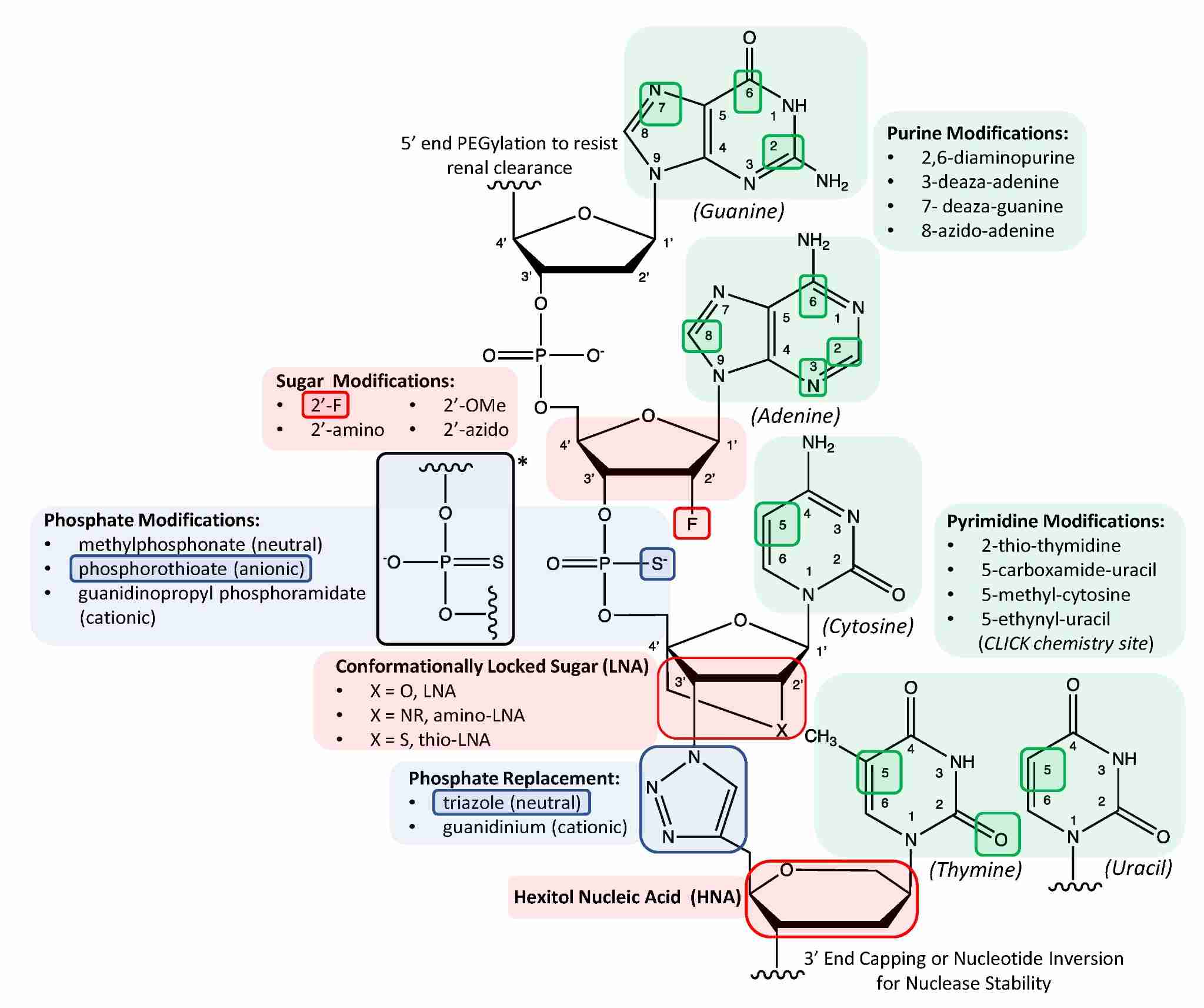 Fig. 2 Schematic illustrating various chemical modifications to the tripartite deoxyribonucleic acid structure ranging from simple site-specific atomic substitutions to more exotic molecular replacements bearing little resemblance to the natural structure.2,5
Fig. 2 Schematic illustrating various chemical modifications to the tripartite deoxyribonucleic acid structure ranging from simple site-specific atomic substitutions to more exotic molecular replacements bearing little resemblance to the natural structure.2,5
Ribose nucleosides have a 2'-hydroxyl that biases toward C-3'-endo sugar pucker and north-conformation helices, features required for RNAi function and ribosomal decoding, but that also makes phosphodiester linkages intrinsically susceptible to base-catalyzed hydrolysis; deoxyribose nucleosides lack 2'-oxygen and bias toward C-2'-endo (south) pucker, leading to DNA strands that are chemically more stable and better recruiters of RNase H. These facts are put to therapeutic use by inserting 2'-O-methyl or 2'-fluoro ribose monomers into the siRNA guide strand to obtain nuclease resistance but A-form geometry, or by inserting locked or constrained-ethyl sugars into the antisense wings to increase melting temperature without negatively affecting RNase H cleavage of the internal DNA gap, a logic-based, sugar-mediated tuning of pharmacokinetics and mechanism of action that can't be achieved with base-only modifications.
Endogenous adenosine, guanosine, cytidine and uridine are relatively unstable as signaling nucleosides (ATP, cAMP, UDP-glucose) as they are cleared by deaminases and phosphorylases; nucleoside analogues such as N1-methyl-pseudouridine or 5-methyl-cytidine are not susceptible to these pathways and thus may have longer half-lives, allowing once-weekly or once-monthly formulations of RNA drugs. C-nucleosides (i.e. pseudouridine) and base-expanded or tricyclic analogues have been introduced that offer C-C glycosidic bonds (chemically stable and not enzymatically recognized) and added H-bond donors/acceptors to fold high-affinity aptamers (thus extending the "chemical alphabet" beyond A, U, C, G), without changing the amidite cycle or automated GMP synthesis lines (enabling next-generation nucleic-acid drugs that are more stable and more specific than native RNAs).
Adenosine, deoxyadenosine is the purine base of nucleic-acid therapeutics, its adenine nucleobase has a large π-surface area for strong base-stacking, and its sugar back-bone can be locked, fluorinated or N-alkylated with no impact on the phosphoramidite coupling cycle, so the same automated synthesizer can produce siRNA, mRNA or antisense drugs whose stability, immunogenicity and target affinity are pre-encoded at the adenosine monomer level — a plug-and-play modularity that shortens lead-time across multiple RNA platforms while retaining a single, regulator-accepted manufacturing backbone.
Adenosine is a purine nucleoside which consists of a bicyclic purine base attached to a β-D-ribofuranose, β-D-2-deoxyribofuranose, or β-D-2-fluoro-ribofuranose via an N-glycosidic bond. The C-6 amino group provides a hydrogen-bond donor for Watson-Crick base pairing and the C-2 and C-8 positions can accept electron-withdrawing substituents to increase base-pairing fidelity without affecting the sugar pucker. Solid-phase synthesis employs the 3'-hydroxyl as its elongation handle while the 5'-hydroxyl receives temporary protection with DMT to enable directional chain growth. These orthogonal functional handles allow iterative amidite coupling to occur without the need for post-synthetic base manipulation, providing a chemically robust backbone for therapeutic oligonucleotides.
Ribose adenosine (A) has a 2'-hydroxyl favoring the C-3'-endo sugar pucker required for RNAi activity, but this also makes the phosphodiester linkage prone to base-catalysed hydrolysis; by contrast, deoxyadenosine (dA) has no 2'-oxygen to bias the equilibrium toward C-2'-endo and so results in DNA strands with increased chemical stability and improved recruitability to RNase H. Gapmer ASOs thus incorporate dA in the middle DNA window to preserve cleavage activity while flanking with 2'-OMe or 2'-F adenosine residues to increase melting temperature and minimize off-target hybridization—all in a single solid-phase synthesis run without any need for post-synthetic sugar substitution.
N6-methyl-adenosine (m6A) is included during in-vitro transcription by T7 RNA polymerase to reduce RIG-I and PKR activation thus improving mRNA half-life without loss of translational fidelity; 2'-fluoro-adenosine improves siRNA guide-strand stability without affecting Argonaute loading, while 7-deaza-8-aza-adenosine improves base-stacking and duplex durability. Modifications are commercially available as phosphoramidites with standard protecting groups so that the same resin can be expected to deliver both gene silencing activity and metabolic stability in a one-pot workflow.
2'-F-adenosine is used at positions 1, 7 and 14 in siRNA duplexes to provide high nuclease resistance but retain seed-region hybridisation, while in mRNA vaccines, N⁶-methyl-adenosine is fed as the triphosphate precursor during T7 transcription to avoid detection by innate sensors and enhance protein expression. Deoxyadenosine is incorporated in the central DNA gap of antisense gapmers to ensure RNase H cleavage, flanked by 2'-OMe-adenosine wings to enhance melting temperature and avoid off-target binding—all using monomer-level encoding and compatible with current GMP synthesis lines.
Thymidine and uridine are the pyrimidine building blocks of DNA and RNA drugs. However, on C-5 thymidine is 5-methylated (enhancing nuclease protection of DNA) and uridine is 5-protonated (yielding conformational dynamics to support ribosomal decoding and siRNA guide-strand positioning); that one atom pre-destines the respective metabolic stability, base-pair conformation and intrinsic-sensor avoidance, and lets the developer interchange T and U (or vice versa) at the monomer level, without changing the solid-phase amidite cycle, and thereby condense multiple drug modalities into a single hardware.
The fundamental function of DNA is information storage. The 5-methyl group on thymidine increases base-stacking energy and sterically shields the double helix from spontaneous deamination, and thereby reduces mutation load during replication. RNA must be able to fold into complex secondary structures and undergo ribozyme or ribosomal catalysis; the 5-proton on uridine favors C3'-endo sugar pucker and north-conformation helices that increase conformational flexibility, a requirement for codon-anticodon wobble and siRNA seed-region hybridization. Therapeutically, this dichotomy allows developers to incorporate T in the DNA core of gapmers in order to preserve RNase H competence while flanking the same oligo with 2'-OMe-U residues to increase melting temperature without using immunogenic base analogues.
Thymidine's C2'-endo ribose configuration flares the major groove and deepens the minor groove which enables DNA binding proteins recognition while uridine's C3'-endo configuration narrows the major groove and adds helical twist which supports RNA-RNA kissing loops and tertiary contacts. The methyl group also makes thymidine a poor substrate for cytidine deaminase and thereby prolongs its plasma half-life, whereas uridine is more easily salvaged and phosphorylated and thereby serves as a fast-acting precursor pool for in-vitro transcription reactions used to manufacture mRNA vaccines. These structural and metabolic differences can be leveraged to engineer DNA-RNA heteroduplexes that confer both the stability of T and functional flexibility of U within a single drug molecule.
Pseudouridine (Ψ) repositions the N1-C1' glycosidic bond to C5-C1', increasing base-stacking and lowering RIG-I recognition; 2-thiouridine provides a soft nucleophile, which augments π-π interactions and blunts TLR7 activation. 5-methyl-uridine (ribothymidine) closely mimics T while preserving RNA sugar pucker. This property can be used to form a "stealth cap" that evades innate sensors without reducing translational efficiency. Both phosphoramidites and triphosphates forms enable solid-phase or enzymatic synthesis of these modifications enabling a single monomer bottle to deliver immune evasion and duplex stability in one workflow process.
DNA gapmers targeting RNAs must have T in the center in order to remain substrates for RNase H. siRNAs, by contrast, use U in the first, seventh, and fourteenth position to better anchor to Argonaute and to permit a flexible seed region. N¹-methyl-Ψ-U is included in mRNA vaccines to prevent PKR activation while maintaining degeneracy. Aptamer libraries are frequently all-ribo except for lack of T. Thus the desired mechanism (DNA backbone for catalysis, RNA for either translation or catalysis, or hybrid for modularity and stability) should determine the decision—and all three can be achieved with the right amidite bottle and without additional synthesis retooling.
Although cytidine and deoxycytidine are used as the pyrimidine "letters" C and dC in RNA and DNA, their exocyclic 4-amino group, C5-H and sugar 2'-OH groups present multiple, orthogonal handles for medicinal editing; methylation, halogenation, sugar locking or arabinose inversion, introduced during solid-phase synthesis, can give rise to antiviral, antitumour or epigenetic drugs that preserve base-pair fidelity but acquire new pharmacodynamic profiles.
The chemical structure of cytidine (ribosyl-cytosine) is a planar aminopyrimidine ring β-N1-linked to ribose, with three vertices available for editing, which are the 4-NH2 (hydrogen-bond donor), C5-H (electrophilic handle) and 2'-OH (conformational switch). In deoxycytidine, the 2'-OH is replaced by hydrogen, which rigidifies the sugar, and allows for greater chemical stability (tolerance to alkaline pH). Both nucleosides have pH-dependent tautomers (lactam ↔ lactim), and are susceptible to spontaneous deamination to uridine/uracil; this instability is used prodrugically. 5-azacytidine irreversibly binds DNA methyl-transferases covalently, while 5-fluorocytosine can be converted by the host to FUTP, which is then lethally incorporated into fungal RNA.
5-methyl-cytidine (m5C) raises Tm by increasing base-stacking energy and does not change Watson-Crick geometry. As such, it is a typical edit used in mRNA vaccines to reduce TLR7 sensing. 5-fluoro-cytosine improves π-stacking and enhances RNase H cleavage specificity, which enables shorter gapmer designs. 2-thio-cytidine places a soft nucleophile, which increases affinity for G while retaining C3'-endo pucker, in aptamer stems. N4-acetyl-cytidine reduces protonation at physiological pH, which lowers non-specific electrostatic interactions in serum. These edits can be introduced during phosphoramidite coupling and so can be incorporated, one pot, into long oligonucleotides without protecting-group gymnastics.
2'-O-methyl-cytidine is incorporated within the passenger strand to mediate TLR7 silencing but is left unmodified in the seed to maintain argonaute contacts; 5-methyl-cytidine is also at position 14 to further stabilize the guide–target duplex and minimize off-target windows. gapmer antisense: a central stretch of deoxycytidines acts as the RNase-H recognition floor, while the 2'-MOE-5-methyl-cytidine wings at either side to increase binding affinity and inhibit cytidine deaminase. Bicyclic cytidine crosslinked with a 4'-CH2–O–2' bridge (LNA-C) is incorporated at each end of the strand to resist exonucleases without causing hepatic offload. Thus, cytidine modifications are used to tune the interplay between catalytic degradation, innate immune silence and PK durability.
Synthesis on a multigram scale requires an efficient procedure without chromatographic intermediate purifications. A convenient route starts from the mild Lewis acid-catalysed protected cytosine nucleobase alkylation, Vorbrüggen coupling to ribose or 2-deoxyribose peracetates, isolation of crystalline β-anomers on controlled cooling. Protection of the N4 position of cytidine monomers must survive the repeated acid cycles (DCA/TCA), but must be removable by mild methanolic ammonia to avoid transamidation. The 5-iodo-cytidine intermediate requires heavy-metal scavenging (Pd, Cu) for injectable material (< 10 ppm). Stereocontrolled introduction of a 5-alkynyl or 5-fluoro group by palladium-catalysed cross-coupling uses ethanol–water, avoiding chlorinated solvents. Final phosphoramidite preparation requires anhydrous acetonitrile and molecular-sieve drying to avoid phosphodiester side-products for > 98 % stepwise yield in 200-mer synthesis. The supply chains for cytidine monomers therefore need orthogonal protection methods, reliable scavenging of metals, and green-solvent methodologies to scale up bench chemistry to GMP tonnage quantities.
Guanosine nucleosides form the molecular backbone of most programmable gene drugs: its bicyclic purine framework features three hydrogen bond donors and two acceptors to seal G–C pairs with strongest affinity, while the C8, N2 and O6 positions provide orthogonal handles for syn/anti locking, lipid conjugation or G-quartet templating, allowing a single monomer to serve duplex tightening, quadruplex assembly or innate immune silencing in siRNA, mRNA and aptamer designs.
Guanine has a flat, aromatic purine ring with hydrogen bond donors at N1 and O6 and an acceptor at N7, allowing canonical Watson–Crick pairing with cytosine as well as non-canonical Hoogsteen base pairing for use in G-quadruplex structures. As such it is used in therapeutics both as the complement in high-affinity RNA duplexes or as a template for self-assembly of G-tetrad scaffolds that can be stabilized with K+ ions in biological environments. In addition to sugar hydroxyls at C2' and C3', the exocyclic amino group at N2 is frequently acylated to transiently block potential transamination reactions during solid phase synthesis, allowing the same resin to encode both gene silencing and metabolic stability in a one-pot process.
8-aza-guanosine shifts the electron-withdrawal of the purine ring to increase base-stacking and duplex melting temperature without perturbing sugar pucker; 7-deaza-guanosine ablates the minor-groove hydrogen-bond acceptor, abrogating RIG-I recognition and prolonging mRNA half-life. The small monophosphates cyclic guanosine monophosphate (cGMP) or deoxy-guanosine monophosphate (dGMP) can serve as "fill-in" ligands that occupy empty G-tetrad sites and provide a metabolite-based anchor for small-molecule conjugates that stabilise telomeric or promoter G4 structures involved in the regulation of oncogenes.
The poor solubility of Guanosine in organic solvents and its vulnerability to N7 alkylation present technical challenges in the synthesis of amidites on a multi-gram scale. Temporary protection of O6 as a cyanoethyl group and N2 as an isobutyryl residue helps to avoid side-reactions during the synthesis. Slow crystallization of the final product from a mixture of ethanol and water produces pure 8-aza or 7-deaza derivatives without the need for chromatographic purification. Glycosylation by flow-chemistry under Vorbrüggen conditions greatly reduces anomeric epimerization, and inline FT-IR monitoring of the product stream can be used to verify complete deprotection before the solution leaves the reactor. Both steps are in line with GMP requirements for impurity profiling and also reduce the amount of solvent used by more than 80 %.
G-rich elements fold into G-quadruplexes or G-ribbons that can be either activating or deactivating; incorporating 2'-O-methyl-guanosine at strategic positions locks the RNA hairpin needed for anchoring the siRNA guide-strand, whereas 2'-fluoro-guanosine stabilizes the aptamer loop required for high-affinity protein binding. On the other hand, designed G-vacancy sites can be introduced to yield "switch-off" oligos that are only reactivated when metabolites from the cellular guanosine pool come to fill the tetrad, providing a metabolite-sensitive on/off switch that couples drug activity to intracellular purine metabolism.
The nucleosides themselves are the alphabet used to encode oligonucleotide drugs, but in their native phosphodiester form they are cleaved by serum nucleases in minutes and are ligands for innate sensors. Programmatically substituting sugar, base and backbone atoms, added as either amidite or triphosphate monomers, turns each letter into a therapeutic glyph, resistant to catabolism and recognition by the immune system while still retaining Watson-Crick geometry, allowing the same automated synthesizer to produce siRNA, mRNA or antisense drug candidates whose pharmacokinetics are encoded at the monomer level.
Table 2 Monomer-level modification matrix
| Modification class | Example | Functional gain | Analytical focus |
| Sugar 2'-OMe | A/C/G/U | Nuclease resistance | Duplex Tm |
| Sugar 2'-F | A/C/G/U | High-affinity wings | RNase H protection |
| Base 5-Me-C | C | Immune evasion | LC-MS/MS ratio |
| Backbone PS | A/T/C/G | Protein binding | Organ distribution |
The small lipophilic 2'-O-methyl (2'-OMe) locks the ribose into a C3'-endo (north) pucker and increases melting temperature by 1–2 °C per residue, as well as masking the phosphodiester and protecting from endonucleases without affecting Argonaute loading for siRNA guide strands. 2'-fluoro (2'-F) swaps out the hydroxyl with an electron-withdrawing group, further stiffening the sugar, increasing RNA affinity and is also compatible with RNase H recruitment if incorporated into gapmer wings. 2'-O-methoxyethyl (2'-MOE) lengthens the alkyl chain, adding a hydrophobic shell which dramatically decreases renal clearance and hepatotoxicity while restoring affinity lost from first-generation phosphorothioate backbones; these sugar modifications are available as standard amidites, allowing for mixed-mode oligos that have 2'-MOE or 2'-F wings flanking a central DNA or 2'-F window respectively, to combine catalytic cleavage with metabolic stability in a single solid-phase synthesis.
5-methylcytidine (5-Me-C) mimics thymidine sterics, increasing base-stacking and decreasing RIG-I/TLR7 recognition, while preserving Watson-Crick geometry--an edit with big impact in mRNA vaccines, where it can simultaneously increase protein expression and decrease activation of innate sensors . N4-acetyl-cytosine or 7-deaza-guanine remove minor-groove hydrogen-bond donors, further reducing immunogenicity without loss of target affinity. Base-expanded analogues such as G-clamps (tricyclic cytosine) add a fourth hydrogen bond, increasing melting temperature by 3–4 °C per residue, and allowing the use of shorter, more drug-like oligos with picomolar potency. Because these base edits are compatible with standard protecting groups (benzoyl, isobutyryl), they slot seamlessly into automated synthesis, allowing developers to encode both potency and stealth into the primary sequence before scale-up begins.
Ideally, these effects are balanced between excessive stabilization (resulting in compromised catalytic cleavage or ribosomal decoding activity) and insufficient stabilization (resulting in rapid renal clearance). Alternating among different amidites (i.e. 2'-MOE "wings" on either side of a 2'-deoxy "window") can restore RNase H activity while also providing days- to weeks-long serum half-lives. Phosphorothioate (PS) backbone substitution can increase protein binding and tissue uptake. However, highly PS-substituted oligos have increased potential to elicit pro-inflammatory effects. Therefore, contemporary design strategies are based on stereocontrolled PS insertions or chimeric PO/PS motifs to achieve nuclease resistance without inducing systemic cytokine induction. Inline monitoring of the manufacturing steps (UV, MS) provide the exact stoichiometry for each modification (exclusion of batch heterogeneity) to ensure the final oligonucleotide is behaving as a single molecular entity (meeting regulatory requirements of batch-to-batch consistency) with the desired pharmacologic profile.
Nucleoside derivatives have to conform to a layered quality envelope starting from the raw material traceability all the way through the product stability: since an amidite impurity or a last minute water drop may translate into oligonucleotide truncations or depurination events during the solid-phase synthesis, each nucleoside is considered a new chemical entity and comes with purity, moisture, elemental impurity and stereochemical specifications commensurate with the ICH Q3D, Q6A and Q11 guidelines, that should also be compatible with continuous-flow amidite chemistry and real time spectroscopic feedback loops.
Each nucleoside is produced by Vorbrüggen glycosylation and selective crystallization; inline FT-IR analysis of the disappearance of the silylated base and the appearance of the β-anomer is used for process monitoring. Offline UHPLC-CAD provides quantification of the regio-isomers, α-anomer and the amount of residual protecting groups (≤0.5 %). Because a single truncated amidite can be the oligonucleotide deletion seeding impurity, purge factors are determined for process-related impurities (silyl reagents, fluoride salts) and proven via spiking studies to ensure trace levels of aldehydes, nitrosamines or other relevant impurities are removed to sub-ppm levels prior to the monomer entering the amidite conversion step.
Nucleosides are hygroscopic, and the presence of residual water in the samples hydrolyses phosphoramidites to phosphoric acid over time during long-term storage. This causes a reduced coupling efficiency and oligonucleotide truncations. To prevent this, crystallized nucleosides are therefore dried under vacuum at ≤40 °C to a water content determined by Karl-Fischer titration to ≤0.2 %, after which the nucleosides are nitrogen packed with a desiccant of silica and real-time head-space moisture sensors on the packaging line are set to automatically initiate re-drying of the material if a humidity greater than 50 ppm is detected. The re-drying process is then followed by accelerated stability studies (40 °C/75 % RH, 6 months) to ensure that anomeric purity and water content are still within specification, and thereby enables an increase in shelf-life to ≥24 months without cold-chain logistics.
Identity is established by 1H-NMR with selective NOE to confirm β-anomeric configuration. Orthogonal 13C-NMR and LC-ESI-MS data are provided to further confirm base and sugar integrity. Residual-metal screening is by ICP-MS with online dilution to give part-per-billion sensitivities for Pd, Sn and Ru catalysts. A forced-degradation matrix (acid/base/peroxide/photolysis) is used to develop a stability-indicating method whose peak resolutions are locked in by retention-time modelling. All data are uploaded to a LIMS that automatically creates batch records, certificates of analysis and regulatory-compliant Module 3 sections, thereby meeting both FDA and EMA requirements for traceability and comparability with no manual transcription.
We provide a comprehensive and technically robust portfolio of A, T/U, C, and G nucleoside derivatives to support the full spectrum of RNA- and DNA-based therapeutic development. Our offerings are designed to meet the evolving requirements of oligonucleotide chemistry, from early discovery to large-scale manufacturing, while ensuring consistent quality, performance, and regulatory readiness.
Our nucleoside derivative portfolio covers all canonical bases—adenosine (A), thymidine or uridine (T/U), cytidine (C), and guanosine (G)—in both ribose and deoxyribose forms. This enables seamless support for RNA therapeutics, including siRNA and mRNA, as well as DNA-based oligonucleotides such as antisense and aptamer drugs. These derivatives are manufactured to stringent specifications, with a focus on high chemical purity, controlled impurity profiles, and batch-to-batch consistency. Native nucleosides, protected intermediates, and synthesis-ready derivatives are available to ensure reliable performance during oligonucleotide assembly and downstream processing. By offering a complete and balanced set of nucleoside building blocks, we help developers maintain consistency across sequences and therapeutic platforms.
To support advanced oligonucleotide design, we offer a broad range of modified and protected nucleosides optimized for chemical synthesis. Our portfolio includes nucleosides with base and sugar modifications as well as carefully selected protecting group strategies that enable precise, stepwise chain assembly. These modified and protected nucleosides are designed to deliver high coupling efficiency, minimal side reactions, and clean deprotection behavior, which are critical for achieving high yields and sequence fidelity. They are compatible with standard solid-phase oligonucleotide synthesis workflows and support the development of therapeutics with enhanced stability, binding affinity, and pharmacological performance.
In addition to standard offerings, we provide custom nucleoside synthesis and scale-up support for programs with specialized requirements. This includes the development of novel nucleoside derivatives, optimization of synthetic routes, and adaptation of protection strategies to accommodate unique base or sugar modifications. Our technical team works closely with customers to ensure that custom nucleoside derivatives are not only chemically sound but also scalable, reproducible, and suitable for long-term supply. This approach helps bridge the gap between early-stage innovation and commercial manufacturing, reducing technical risk and enabling smoother program progression.
If you are seeking reliable, high-quality A, T/U, C, and G nucleoside derivatives supported by experienced technical expertise, we are ready to assist. Contact us today to request technical information, samples, or quotations, or to discuss custom nucleoside synthesis and scale-up solutions tailored to your RNA or DNA therapeutic program.
References
They are chemically modified or protected forms of canonical nucleosides used in RNA and DNA drug synthesis.
Derivatives improve stability, synthesis efficiency, and compatibility with chemical assembly methods.
Yes, but the specific nucleoside (T vs U) depends on whether the drug is DNA- or RNA-based.
Yes, both base and sugar positions can be modified to improve drug properties.
Yes, they influence stability, binding affinity, and overall therapeutic efficacy.


 Loading ......
Loading ......