Raw material decisions made at the onset of process design essentially determine the economic, regulatory, and technical direction of any oligonucleotide program. Inherently, all downstream processing steps, from solid-phase synthesis, cleavage, purification to sterile filtration, reflect the impurity profile and physical properties of the nucleoside phosphoramidites. A suboptimal choice at the gram stage can therefore lead to multi-kilogram yield losses, failed specifications, and ultimately result in costly regulatory revisions. Raw material strategy therefore involves careful evaluation of chemical purity, physical form, the mapping of impurities to their fate, supplier audit rigor, and long-term supply chain security under cGMP-compliant expectations.
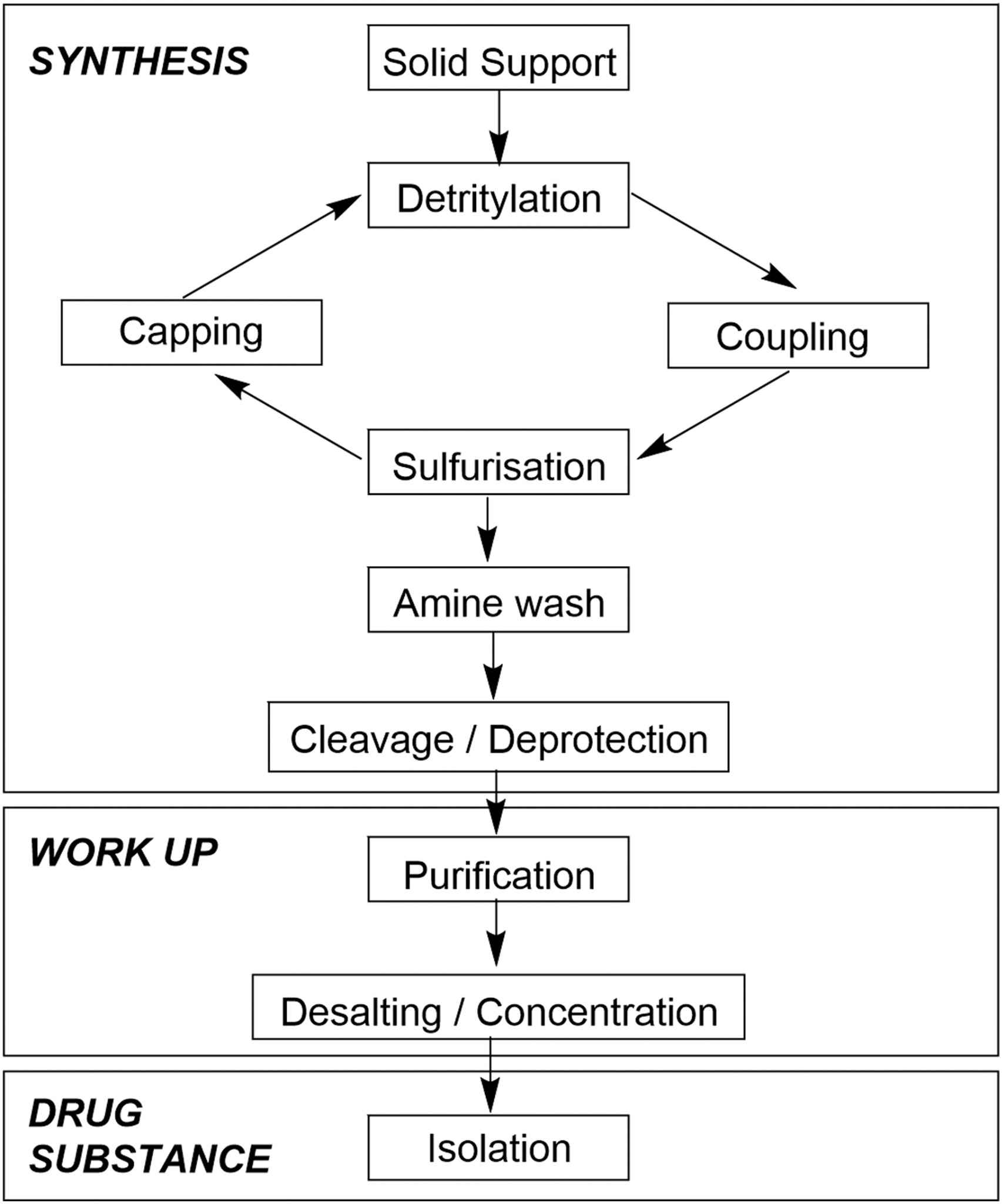 Process overview of oligonucleotide manufacturing operations.1,5
Process overview of oligonucleotide manufacturing operations.1,5
The gap between a laboratory synthesis that scales to milligrams and a commercial campaign that reliably delivers kilograms is determined primarily by the quality, consistency and traceability of the incoming nucleoside building blocks. Adopting scalable, well-characterized raw materials early on averts expensive re-validation, reduces solvent usage per gram of API and maintains synthetic yield when column volumes, reactor sizes and cycle times are pushed to the plant limits. Late-stage substitution of a "research-grade" phosphoramidite forces repeat tox lots, revised specifications and regulatory filings that can eat up months of launch schedule.
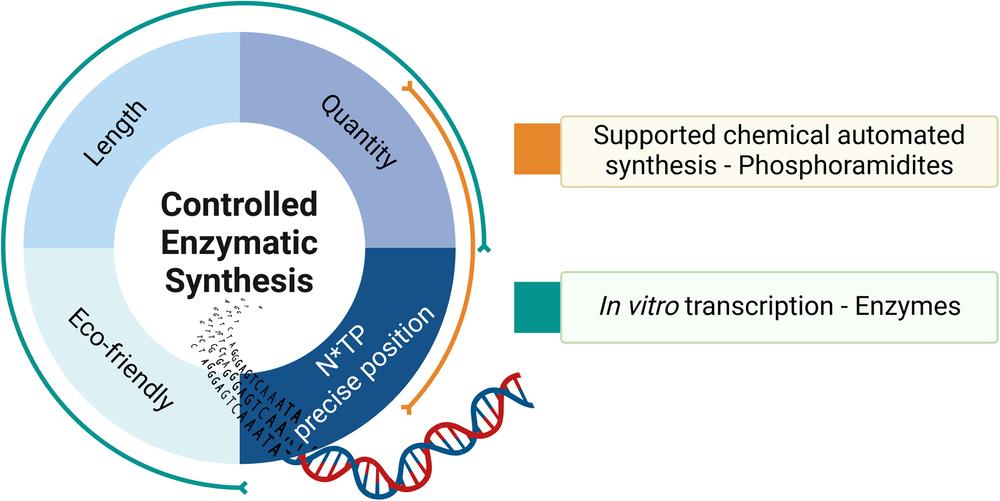 Pros and cons of chemical automated synthesis and enzymatic synthesis of oligonucleotides.2,5
Pros and cons of chemical automated synthesis and enzymatic synthesis of oligonucleotides.2,5
Chromatographically silent impurities at 1 mmol scale become chain-terminating or mutagenic after thousands of coupling cycles. Trace moisture, residual metals or abnormal anomeric ratios lead to reduced coupling efficiency, shorter fragments and increased load on downstream ion-exchange or desalting steps. A nucleoside lot passing "nuclease-free" specs that contains 0.1% free nucleobase, for example, can give rise to a hydrophobic adduct that co-elutes with the full-length oligonucleotide, requiring repeated preparative injections and yield loss. Non-negotiable for scale-up is therefore very rigorous release testing—identity, water content, anomeric purity, residual solvent, trace metals, bioburden—and an accompanying unambiguous impurity fate map.
At bench scale a failed synthesis is an irritation; at ton scale it is a plant shutdown. Phosphoramidites acceptable at the research-grade level come with little documentation but commercial campaigns require supplier audits, change-control agreements, dual sourcing and validated analytical methods that identify and track every impurity to its source. Physical properties change as well: powders must flow, not cake, when transferred under vacuum; moisture specification is tighter, because a 50 ppm increase can cut the coupling efficiency in half when 100 kg of amidite is dissolved in 800 L of anhydrous acetonitrile; particle size distribution is important, as dusting of powders can plug fluid-bed dryers. Finally, solvent volumes per kilogram of oligonucleotide decrease by an order of magnitude, so impurity amplification factors increase commensurately.
Changing the nucleoside supplier following key tox or Phase II lots has major implications: side-by-side synthesis, complete analytical re-characterization, stability re-qualification, and usually a fresh regulatory amendment. Even a simple switch—from one crystallization solvent to another—can result in changes to impurity profiles, retention times, or solid state properties that impact dissolution rate and filterability. Expect requests for data to support claims of no difference in potency, safety and immunogenicity profiles; in practice this means multiple pilot batches, extended stability stations, and sometimes a bridging non-clinical study. Financial risk routinely runs 10 times the original savings in raw material costs, leading to the guiding principle that nucleoside choices are best locked in before first-in-human dosing.
Table 1 Comparative selection criteria for research-grade vs. commercial-grade nucleoside phosphoramidites
| Attribute | Research Grade | Commercial Grade |
| Typical release assays | Identity, purity | Identity, purity, water, trace metals, residual solvent, bioburden, particle size |
| Impurity fate map | Not required | Required with toxicological rationale |
| Change control | Minimal | Formal, with regulatory notification |
| Supply agreement | Purchase order | Long-term contract, dual sourcing |
| Physical form | Any workable solid | Free-flowing, low-dust, defined crystal habit |
| Documentation | Certificate of analysis | DMF/CEP available, audit report, stability data |
Chemical purity, lot-to-lot reproducibility, water control and compatibility with protecting-group protocols are the four critical factors that determine if a phosphoramidite works successfully at multi-kilogram scale. Each of these factors has a multiplicative effect: an otherwise uncontrolled trace impurity or a 20 ppm increase in water that may be acceptable at 50 µmol scale can result in metric tons of truncated sequences, thousands of pounds of failed anion-exchange pools, and severe regulatory questioning when column sizes and volumes of acetonitrile are multiplied. The choice of phosphoramidite must be data driven, from a complete understanding of impurity fates to a supply contract that specifies the synthetic route, crystallization solvent, packaging configuration and change-control processes.
Any nucleoside entering a phosphoramidite plant must have an impurity profile that is not only low, but predictably removable or dilute-able through the downstream train. Process-related markers—unreacted sugar acetates, base adducts, anomeric isomers, residual metals from glycosylation—must be individually tracked because each can terminate chain extension, create toxic side-products, or co-elute with the final API during ion-exchange or desalting. Regulators expect a toxicological rationale for every peak above the ICH threshold; consequently, early cross-spiking studies in which putative impurities are deliberately doped into pilot syntheses are essential to demonstrate purge. Purity specifications should therefore be route-specific rather than copied from catalog monographs, and each new supplier or synthetic revision must trigger a fresh impurity re-qualification campaign.
Manufacturing campaigns for commercial oligonucleotides run dozens of consecutive batches on fully automated synthesizers. As little as 2% drift in the reactivity of individual amidites or particle morphology over time is enough to cumulatively alter crude purity profiles enough to overload purification resins and invalidate previously validated pooling rules. Reproducibility is built by putting the entire manufacturing chain under a formal change-control system, starting with the initial base material, sugar protection sequence, crystallization solvent, milling protocol and even final packaging atmosphere. Statistical process control on the most critical attributes, such as anomeric ratio, water uptake rate, and free base content, also provide early warning for vendor drift. Dual sourcing is always encouraged but not before side-by-side equivalence demonstrations that include full pilot-scale syntheses followed by stressed stability studies of the crude drug substance, as well as simple analytical overlay.
Phosphoramidite chemistry is essentially a race between coupling cycle and hydrolysis; the nucleoside must therefore be received with very tight control of bound moisture specification and maintain that level for its labelled shelf-life. Packaging format - aluminum pouches under argon atmosphere, low-permeability fluorinated bottles, or sealed cans with desiccant inserts - must be qualified by real time storage studies that simulate warehouse temperature excursions and not just 25 °C/60% RH comfort zones. As important is the uptake rate once the container is breached; hygroscopic powders that exceed the critical 100 ppm threshold within minutes on a humid plant floor will defeat even the best anhydrous acetonitrile delivery systems. A pragmatic control strategy couples factory-in-use time limits with in-line NIR or Karl-Fischer spot checks so that batches are rejected before they enter the synthesis suite rather than after a failed coupling audit.
The nucleoside will also have to tolerate repeated cycles of acid deprotection, iodine oxidation and base deblock without producing side reactions that can alkylate or depurinate the elongating strand. Steric congestion from 2'-orthoester or 2'-thio protecting groups will also exacerbate poor coupling kinetics if the 3'-phosphoramidite's steric bulk is not well matched to the base, leading to truncated fragments that contribute to purification load. Similarly, base-protecting groups that require long treatment times with methanolic ammonia can also promote transamidation or cytidine deamination if the nucleoside itself contains impurities such as residual Lewis-acidic metal ions. Such issues should be surfaced as early as possible through early screening that "stress-tests" each candidate amidite in a four-base mixed sequence context and with accelerated coupling times and exaggerated exposure periods, not only in terms of stepwise yield, but also in terms of the onset of base-loss or isomer peaks. Successful qualification will be reflected in a documented design space that correlates temperature, coupling time and activator excess with acceptable impurity levels, such that the raw material will remain in its demonstrated acceptable region even when process parameters drift in the plant.
The distinction between "research grade" and "GMP grade" nucleoside monomers is one of degree of documented control; the former is marketed against broad, frequently non-published specifications and the latter is produced within a pharmaceutical quality system that anchors each batch record to validated analytical methods, change-control oversight, and regulatory filings. The reason for this split is not arbitrary: early stage discovery efforts must be agile and milligram flexible, but the manufacture of clinical scale lots must ensure that every kilogram of amidite is manufactured with the same fate of impurities, kinetics of moisture uptake, and trace-metal profile as the lots delivered for toxicology. Switching tiers at the wrong stage of development incurs rework of expensive repeat stability studies, bridging tox batches, and in the worst case a complete re-write of the Drug product specification.
Research-grade monomers are most often provided with a single-page certificate of analysis providing nominal HPLC purity and not much else. Anomeric distribution, residual solvent class, heavy-metal panel, bioburden, and moisture content can be missing or declared as "typical" ranges. GMP-grade material on the other hand will include: synthetic route with key controls identified, validated impurity profiling with pre-defined acceptance criteria, risk assessment for nitrosamine and mutagenic impurities, resin-cleaning verification, container-closure headspace moisture ingress, and stability statement for both long term and in-use conditions. Batch records need to be audit-ready, samples need to be retained and archived for regulator inspection, and even post-approval changes require a formal variation. The goal is not just "high purity" but predictable, traceable, and reproducible purity, in the face of inspectors who might request raw integrals, calibration curves, and system-suitability reports.
While pre-clinical feasibility may begin with research grade amidites legally, there is increasing agency expectation for an early demonstration that the impurity profile will not negatively impact safety pharmacology or add assay artifacts in genotoxicity. After entry into GLP tox, the active pharmaceutical ingredient (API) precursor must be produced under at least an ISO certified quality system and Phase I chemistry must follow the route and controls planned for commercial launch (barring a minor toxicology 'bridging' package). After Phase II all deviations from GMP grade starting materials will need a comparability package of side-by-side syntheses, impurity purge calculations and often a new stability commitment. The unstated message is "research grade is fine when there is no pharmaceutical grade equivalent but when there is the sponsor must submit a risk mitigation package that addresses specification gaps, supply assurance and a transition timeline".
Ideally the transition should have been done before GLP tox begins, since qualification after the fact is orders of magnitude more expensive once you have pivotal animal data in the file. A realistic timetable starts with a gap analysis during lead-optimization: compile a list of all the tests not on the research-grade certificate, send in a small GMP pilot lot and do a side-by-side coupling comparison to demonstrate equivalent crude yield and level of impurities removed, etc. In parallel, audit the supplier's quality system, finalize the synthetic route and place the material on accelerated and long-term stability. If necessary, a phased-in approach can be acceptable - research-grade for first tox and GMP-grade for repeat-dose and reproductive tox - if the bridging data show the latter has equivalent or better purity and no new mutagenic indication. If you leave it until Phase II, you have risked the program on possible supply problems, specification creep, and the risk of having to repeat tox studies under a different impurity profile (this happens all the time and routinely adds 6-9 months of critical-path delay).
A nucleoside route is only considered scalable if the same set of transformations that provides grams in a fume hood can be piped into stirred-tank trains or continuous-flow coils without rewriting the chemistry; every previous decision—solvent, exotherm profile, crystallization solvent, etc.—is locked in when clinical need presents itself, so the process must already have headroom for heat and mass transfer that is not observable until multi-ton scale.
Robustness starts with thermal neutrality: reactions that proceed on a narrow temperature margin in the laboratory will channel or bum once jacket area per volume goes down. Routes are therefore designed with exotherms spread across several elementary steps, each diluted in high-boiling, water-immiscible solvents that can be gravity-settled rather than centrifuged. Protecting-group manoeuvres are minimized to avoid sequential high-vacuum strip-and-re-dissolve cycles that quickly become batch-time bottlenecks; instead, telescoped washes move crude intermediates forward as slurries. Work-up must tolerate inadvertent overcharge of acid or base because reagent metering accuracy drifts at plant scale; buffers or latent bases are engineered in so that a 20% excess still lands inside the validated pH corridor. Finally, every solid phase is filtered on a Buchner during development to ensure that the same cake compressibility allows leaf-filtration or centrifuge discharge later.
A pathway dependent on a single-source sugar or custom heterocycle takes on all of that upstream geopolitical, harvest, or regulatory risk. Scalable processes therefore use commodity feedstocks (anhydrous ribose, protected cytosine, pyridinium tosylate) traded under multi-supplier contracts with published specifications. Where a proprietary intermediate is unavoidable, the developer either negotiates a technology-transfer pack (analytical methods, starting-material specs, change-control calendar) or commissions a second supplier to duplicate the sequence using orthogonal protecting groups so that any force-majeure event can be mitigated by rerouting rather than by revalidating the entire synthetic tree.
Table 2 Supply-Risk Mitigation Matrix
| Intermediate Tier | Risk Level | Typical Mitigation Approach |
| Commodity carbohydrate | Low | Dual-source contracts, annual price lock |
| Halogenated heterocycle | Medium | Second supplier with different route |
| Chiral auxiliary | High | In-house stockpile, captive production clause |
| Catalyst ligand | High | Route redesign to abolish need |
Yield is therefore always a time-averaged number over the life of a campaign: 90% isolated yield dropping to 70% after three solvent-recycle loops is more margin-dilutive than 85% overall yield that remains consistent. Developers optimise for robustness rather than absolute numbers, accepting a greater number of steps if each step operates within a wide design space. Cost modelling includes solvent losses to turnover, energy costs for cryogenic grinding and carbon-credit exposure for chlorinated waste; the reagent that appears cheapest on paper can become the most expensive when incineration surcharges are factored in. Supply-chain resilience is tested by simulating a six-month port closure: only supply routes whose critical raw materials can be air-freighted without cold-chain or hazardous-class restrictions pass the exercise.
Capacity planning, therefore, matches forecast oligonucleotide demand to reactor volume, not nominal throughput. As phosphoramidite monomers are used in molar excess during solid-phase synthesis, demand for a given nucleoside may vary by a factor of ten depending on the coupling excess chosen for a therapeutic candidate. Developers therefore negotiate stepped supply agreements that reserve campaign slots on 10 m³ reactors with the option to call down additional trains as Phase II enrollment accelerates. Conversely, capital-light virtual manufacturing is preferred early on: the supplier retains ownership of fixed assets while the developer books variable volume, converting to dedicated lines only once revenue predictability justifies the balance-sheet drag. Regulatory commitments are matched in parallel—additional supplier audits, extractable studies on new hoses, and stability chambers sized for double the forecasted annual launch volume—to prevent a surprise approval surge from colliding with capped warehouse space.
Control of a nucleoside monomer does not really exist until the strategy behind it can demonstrate with statistical certainty that each subsequent lot will possess the same fingerprint of identity, impurity profile, residual-solvent spectrum and moisture load as the one that entered toxicology. That requires an integrated analytical approach – identity verified by orthogonal spectroscopy, purity by multi-detector chromatography, metals by ICP-MS, volatiles by head-space GC, water by Karl-Fischer coulometry – that is locked into a change-control system that treats each new vial as an extension of, not a re-identification of, the previous batch. The sections that follow unpack the non-negotiable pillars: profiling strategy, trace-element control, documentation spine and supplier technical responsiveness.
Identity needs to be proven by at least two complementary techniques, generally proton/carbon NMR plus high-resolution MS, so that structural isomers or anomeric variants cannot go undetected when scale increases their concentration. Purity is established by a primary HPLC-UV method fully validated for specificity, linearity and limit of detection, with an orthogonal secondary separation (ion-pair or hydrophilic interaction) to force resolution of co-elutors not visible to a single stationary phase. Impurity profiling is route-specific: each synthetic intermediate, side product or forced-degradant is spiked into the matrix to establish relative response factors so that later "new" peaks are immediately flagged rather than dismissed as baseline noise. A living impurity library, updated whenever the synthetic route or crystallization solvent changes, provides the toxicological rationale required by ICH Q3A and precludes last-minute surprises at regulatory review.
Head-space GC with electron-capture or mass-selective detection is used to determine the amount of each Class 2 and Class 3 solvent used or formed during sugar protection, base acylation and phosphitylation; Class 1 solvents are required to be shown to be absent at the limit of detection. Metals introduced through catalysts, filter aids or stainless-steel transfer lines are profiled using ICP-MS operated in collision-cell mode to resolve argon-based polyatomic overlaps; the panel includes not only the required ICH Q3D oral/IV option, but also extends to catalyst specific elements (e.g., Pd, Ru, Rh) where acceptance criteria is based on the allowed daily exposure instead of arbitrary ppm limits. Moisture is measured by Karl-Fischer coulometry performed in an inert gas environment because even trace water will hydrolyse phosphoramidite moieties during long-term storage; the method is validated for both closed-vial and in-use sampling to represent factory floor exposure. Taken together, these three tests comprise the "hidden mass balance" that can be used to close the assay-purity gap that is often seen during technology transfer.
Each analytical method is maintained in a strict document lineage: method development report, qualification summary, validation protocol, and finally the routine SOP. Each change – column lot, internal standard supplier, gradient adjustment – is the subject of a formal deviation investigation, and if significant, a re-validation exercise. Batch traceability is ensured by a chain-of-custody log that documents the relationships between the starting nucleoside batch, the phosphoramidite lot used, the cleaved crude drug substance, and the final formulated vial. The complete pedigree can be rapidly reviewed if a OOS result is observed months after release. Reference standards are qualified following WHO-style guidelines: identity confirmed by NMR, purity checked by quantitative NMR and HPLC-UV, potency corrected for residual water, counter-ion, and inorganic content, and stored in qualified refrigeration with re-test dates. Electronic data systems must be 21 CFR Part 11 compliant with audit-trails on chromatographic integrations, spectral overlays, and calculation spreadsheets.
A credible supplier provides a technical package—not just a certificate. It is an easily-accessed package of forced-degradation spectra, impurity fate and disposition stories, cross-spiking studies and above all a commitment to meet in a pre-specified time window for out-of-specification events. When an unexplained HPLC shoulder at 2 °C appears six months into commercial manufacturing, the vendor can readily share historical stress data, offer to modify the column, or provide an orthogonal UHPLC method in days not weeks. On-site analytical audits are supported by round robin testing in which the same batch is analyzed in both the vendor and customer laboratories to rule out systematic bias. Finally a living analytical development roadmap with planned future work to increase method robustness, monitor new genotoxic impurity alerts, or upcoming pharmacopoeial monograph revisions helps the developer to be proactive in anticipating regulatory requirements instead of reacting to deficiency letters.
Developers consistently underestimate the rate at which a "high-purity" catalog monomer becomes a scale-up liability when multi-kilogram oligonucleotide campaigns are initiated. The most common pitfalls—neglecting heat- and mass-transfer realities, accepting spartan documentation, and sourcing from a single supplier—are subtle because they do not become apparent during milligram route scouting; they rear their heads only when cGMP auditors, process engineers and downstream purification teams become involved. Recognizing and avoiding these pitfalls early obviates the expensive process of repeat tox lots, failed validation batches and emergency supplier changes that can erase months of launch schedule.
Laboratory routes that involve cryogenic lithiations, silica-gel plugs or high-vacuum distillation will often become plant bottlenecks where jacket cooling capacity, column volume availability or solvent-recovery yield are rate-limiting. A nucleoside protection sequence that works smoothly at 0.1 mol scale can stall at 20 mol because viscous slurries prevent efficient agitation, or because exothermic deprotection outstrips heat removal and causes base decomposition that requires a chromatographic rescue that erodes yield and solvent budget. Early techno-economic screening—matching reaction enthalpy to plant heat-transfer coefficients and verifying that each intermediate can be isolated by filtration or extraction rather than chromatography—can expose these constraints while route changes remain inexpensive. Equally critical is the fate of side-products: an impurity innocuous at 0.5% w/w may accumulate in mother liquors, saturate solvent recovery streams and eventually co-precipitate with the final amidite, forcing costly re-work campaigns that were never contemplated during discovery.
Research chemicals are often provided with a one page certificate stating the nominal HPLC purity and a general storage recommendation; brevity is tolerable in a research context but such a file is unsustainable once a regulatory assessor requires a complete impurity disposition narrative, residual-metal rationale and both long-term and in-use stability data. Many gaps are invariably found when a filing team is asked to back-assemble these files at the eleventh hour- no analytical validation for certain methods, a solvent change for crystallization never recorded, supplier audit reports missing etc., all of which can result in repeat tox studies and bridging stability work. It is therefore imperative that a controlled change policy is in place and initiated no later than the first GLP synthesis: all synthetic steps, work-up solvents and analytical methods are put under version control, and any variation (even a new lot of filter aid) results in a formal deviation investigation and an updated impurity profile. Lack of this continuously updated documentation backbone is the reason some developers receive deficiency letters that interrogate the equivalence of the material that was used for toxicology versus commercial manufacture, which can add 6-9 months of regulatory delay.
Dependence on a single source for a safeguarded sugar or iodinated core is an existential risk. An accident at the pilot plant of a contract manufacturer, a contamination incident with the starting materials or, worst of all, a politically motivated export restriction has the power to bring an oligonucleotide programme to a standstill within a matter of days. Double-sourcing is more than a purchasing requirement: technically, the secondary manufacturer must be using the same synthetic route, and produce the same impurities. Both sources must also agree on analytical methods, so that the alternative batches can be used interchangeably in the validated coupling procedures with no impact on yield or crude purity. Mature developers evaluate backup suppliers early in development, qualify the intermediate with them in parallel pilot syntheses, and extend framework agreements for the full commercial campaign well in advance. When a second source is not available, companies are building manufacturing route diversity (enzymatic transglycosylation, continuous-flow halogenation) or strategic inventory buffers of at least one commercial batch size.
Table 3 Common pitfalls and preventive tactics in nucleoside raw-material selection
| Pitfall | Early Warning Signal | Preventive Tactic |
| Cryogenic or chromatography-heavy route | Heat-transfer mismatch at >100 L | Run calorimetry and solvent-recovery mass balance early |
| One-page certificate only | Missing impurity fate data | Demand full analytical dossier before GLP tox |
| Single supplier | No audited backup on file | Qualify second source with side-by-side synthesis |
| Unrecorded solvent switch | New unknown peak in stability pull | Lock synthetic route under formal change control |
| High-dust or hygroscopic solid | Flow stoppage during pneumatic transfer | Specify particle-size band and moisture uptake kinetics |
We provide nucleoside raw material solutions designed to support scalable and robust oligonucleotide manufacturing. By combining high-purity materials, regulatory-aware quality systems, and long-term supply capabilities, we help customers mitigate manufacturing risk and maintain consistency as programs advance from development to commercial production.
Our portfolio includes a comprehensive range of high-purity native and modified nucleosides suitable for RNA- and DNA-based oligonucleotide therapeutics. These materials are manufactured under strict quality controls to ensure consistent purity, low moisture content, and tightly controlled impurity profiles. High-purity nucleosides are essential for reliable phosphoramidite preparation and efficient solid-phase synthesis. By minimizing variability at the raw material level, our nucleosides support predictable coupling efficiency, improved yields, and consistent final product quality, particularly in large-scale manufacturing.
We offer GMP-ready nucleoside supply options designed to meet the regulatory expectations of clinical and commercial oligonucleotide manufacturing. Our materials are supported by comprehensive documentation, including specifications, certificates of analysis, traceability records, and change control processes. Our quality systems are structured to support regulatory submissions and audits, helping customers align raw material selection with long-term compliance requirements. This regulatory-focused approach reduces the risk of late-stage material changes and supports smoother transitions into GMP production.
For programs requiring specialized or non-standard nucleoside derivatives, we provide custom synthesis and scale-up expertise. Our technical team supports synthetic route development, process optimization, and impurity control to ensure that custom nucleosides are robust, reproducible, and scalable. By addressing scale-up considerations early in development, we help customers avoid common manufacturing challenges and ensure that custom raw materials remain viable as demand increases.
Reliable long-term supply is critical for successful oligonucleotide manufacturing. We work with customers to establish long-term supply partnerships that prioritize continuity, capacity planning, and consistent quality. Through proactive communication and supply planning, we help ensure that nucleoside raw materials remain available throughout the development lifecycle, supporting uninterrupted manufacturing and long-term program success.
If you are seeking reliable, scalable nucleoside raw material supply for oligonucleotide manufacturing, our team is ready to support your program. Contact us today to discuss your manufacturing needs, request technical documentation or samples, or explore custom synthesis and long-term supply solutions tailored to your development and commercialization goals.
References
Late-stage changes can cause delays, regulatory risks, and cost increases.
Typically high purity with strict impurity and moisture control.
Before clinical manufacturing and regulatory submission.
Supply interruptions can halt production and delay trials.
Yes, with robust synthetic routes and experienced suppliers.


 Loading ......
Loading ......