Cap 0, Cap 1 and Cap 2 analogs are intermediates of 5'-end methylation that control translational efficiency, immunogenicity and stability of IVT mRNA. Cap 0 carries the N7-methylguanosine triphosphate bridge only, whereas Cap 1 includes an essential 2'-O-methyl modification on the first ribose. Cap 2 is further extended to include the same 2'-O-methyl modification on the second nucleotide. Higher degree of 5'-cap methylation reduces innate immune recognition and increases ribosome binding, thus designing the cap for different IVT mRNA applications including short-term vaccine expression and long-term protein replacement.
The 5' cap is a post-transcriptional, non-templated modification required for in-vitro transcribed mRNA to be biologically active. The 5' cap protects mRNA against 5'-to-3' exonucleases and is a binding site for translation initiation factor complex eIF4E. In synthetic mRNA production, the cap is introduced enzymatically or co-transcriptionally using cap analogs, and the exact chemical structure of the cap immediately determines the mRNA's susceptibility to innate immune sensors, its translational ability in different cell types and its stability in formulation and in vivo.
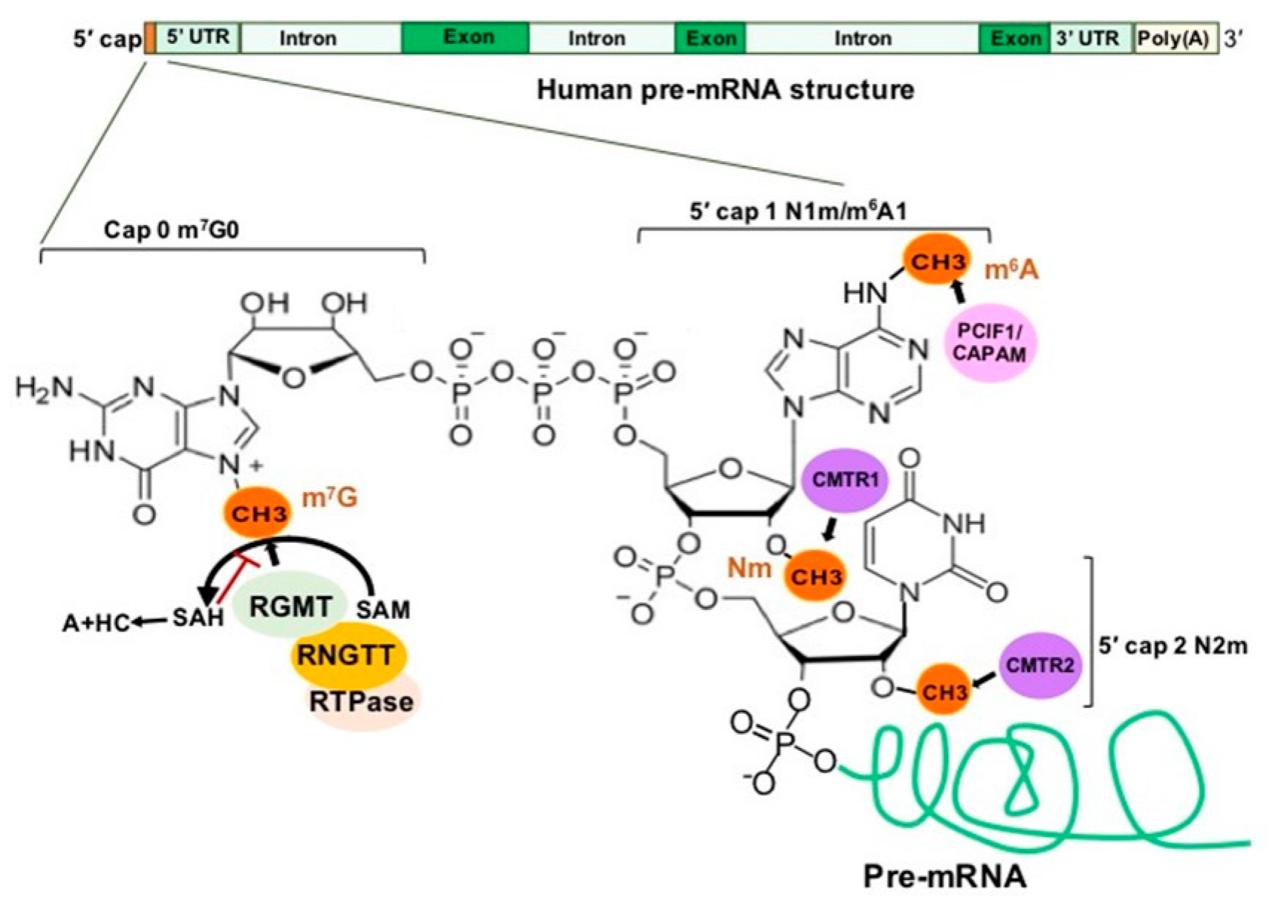 Fig. 1 Cap Structure of a Human Pre-mRNA and the Associated Methylations and Methyltransferase Complexes.1,5
Fig. 1 Cap Structure of a Human Pre-mRNA and the Associated Methylations and Methyltransferase Complexes.1,5
The 5' cap of eukaryotic mRNA is a post-transcriptional modification made of N7-methylguanosine linked to the first ribonucleotide of the primary transcript through a 5'-5' triphosphate bridge. The cap structure, defined as m⁷GpppN, shields the 5' end from decapping enzymes like DCP2 and DXO and acts as a binding site for the translation initiation factor eIF4E. The recruitment of eIF4E recruits the 43S pre-initiation complex and licenses ribosome scanning. However, RNAs lacking a cap or a methylated cap are recognized as foreign by cytosolic pattern-recognition receptors like IFIT1. IFIT1 competes with eIF4E for cap binding and induces translational arrest and RNase L-mediated degradation. The cap of endogenous transcripts is often further modified with 2'-O-methyl groups on the first and second nucleotide to form structures identified as self RNA. For in-vitro transcription, a cap analog, for example, CleanCap AG is added to mimic these natural structures so that synthetic mRNA can avoid the innate immune system and drive efficient protein expression.
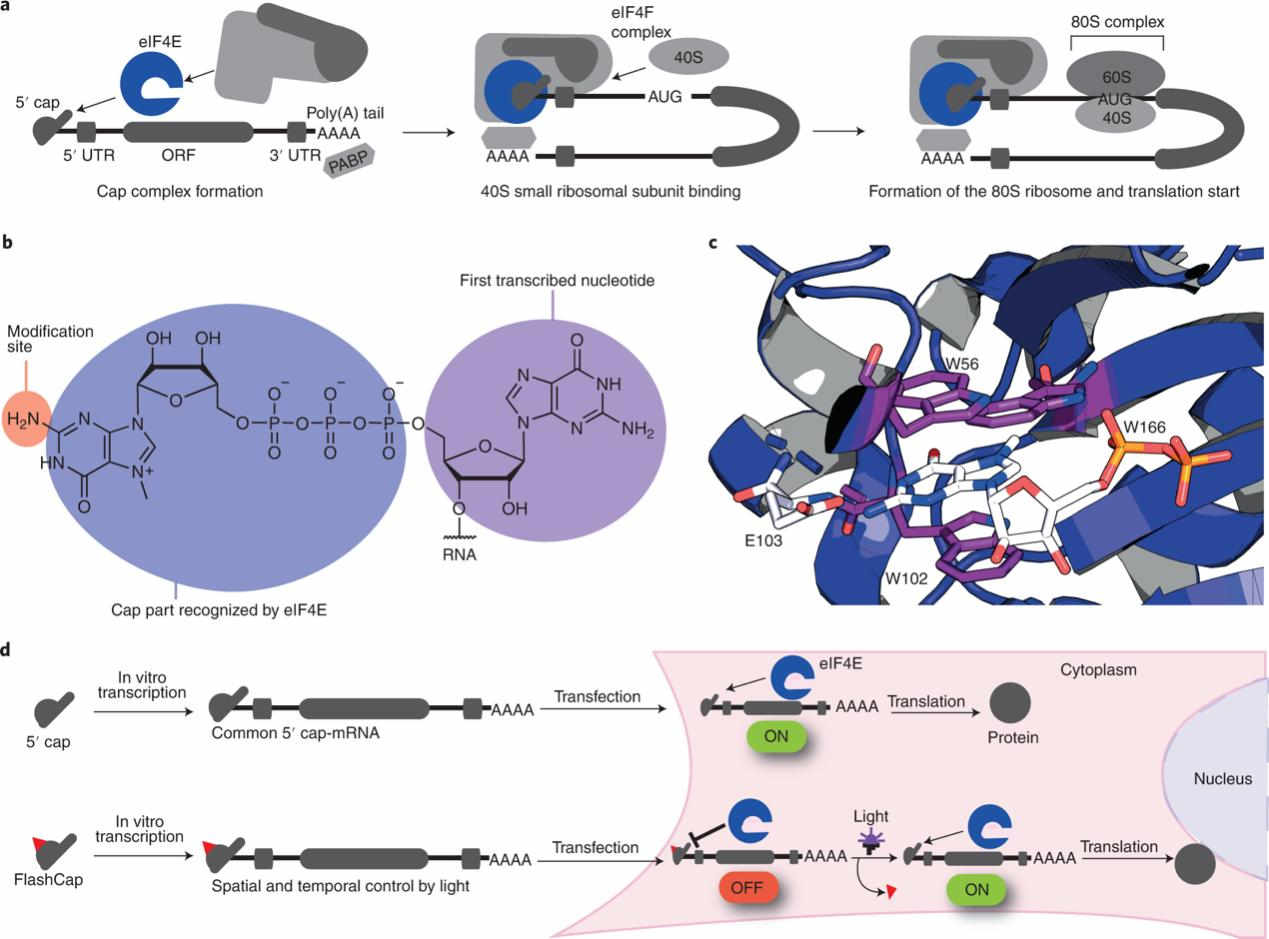 Fig. 2 The 5' cap is a hallmark of eukaryotic mRNAs governing translation initiation.2,5
Fig. 2 The 5' cap is a hallmark of eukaryotic mRNAs governing translation initiation.2,5
The choice of cap structure will directly impact whether IVTT-generated mRNA will be efficiently translated or instead recognised as pathogen-associated molecular pattern (PAMP) that will trigger an innate immune cascade, culminating in silencing of protein expression. Cap 0, which contains no 2'-O-methylation (Nm), is a substrate for IFIT1, which can recognise the cap and directly sterically block eIF4E binding, priming translational arrest and RNase L-mediated mRNA degradation. As a result, Cap 0 mRNAs are immunogenic and therefore not ideal for use as prophylactic vaccines. However, this immunogenicity may be harnessed in a therapeutic setting such as in cancer immunotherapy. Cap 1, which is modified with a methyl group on the 2'-hydroxyl group of the first nucleotide in the mRNA, is not recognised by IFIT1 and so is deemed a signature of self RNA. It has been shown to improve translation by five- to ten-fold and reduce interferon induction. Cap 2, in which the second nucleotide is also methylated, confers further protection against DXO, an interferon-inducible decapping enzyme. This additional protection against decapping would therefore be expected to result in a longer half-life in inflammatory micro-environments. As a result, the choice of cap analog not only impacts on manufacturing strategy but also regulatory acceptance with Cap 1 being the best compromise for most use cases.
Cap 0, Cap 1, and Cap 2 are a series of 5' cap structures that only differ in the extent of 2'-O-methylation of the first and second nucleotide, and form a gradient of immunogenicity and translational efficiency. Cap 0 (m⁷GpppN) (the most basic cap structure) is the structure formed when guanosine cap analogs are used during in-vitro transcription (and has the lowest amount of protection from exonucleases, and will be recognized by IFIT1) and thus is mainly useful for in vitro study where activation of the immune system is needed or wanted. Cap 1 has a single 2'-O-methyl group on the first nucleotide. This modification prevents recognition by IFIT1 and is the cap structure that is most efficiently installed by the vaccinia capping enzyme. Cap 2 has a further methylation on the second nucleotide, which offers extra protection from DXO mediated decapping and exonucleolysis, and is useful in the context of IFN rich environments (eg. tumors, inflamed tissue etc.).
Table 1 Comparison of cap analog structures and functional attributes.
| Cap type | Core structure | 2'-O-methylation | Primary advantage | Typical limitation |
| Cap 0 | N7-methyl-GpppN | None | Basic nuclease protection | Innate immune activation |
| Cap 1 | N7-methyl-GpppN | First nucleotide | Reduced TLR7/8 signalling | May still activate PKR |
| Cap 2 | N7-methyl-GpppN | First and second nucleotide | Maximal immune evasion | Synthesis complexity |
The organization from Cap 0 to Cap 2 is characterized by an increasing number of methyl groups on 2'-hydroxyl group of the first two ribonucleotides, with the 5' terminus of the molecule going from having a basic cap to a complete mature self-mark that can circumvent various checkpoints of the innate immune system. With each methylation, the susceptibility to decapping and inhibitory translational arrest by interferon signaling becomes lower and lower, providing a spectrum of sensitivity that can be calibrated to the clinical needs.
Cap 0 analogs are composed of N7-methylguanosine joined to the first nucleotide of the transcript by a 5'-5' triphosphate bridge. The resulting structure is abbreviated m⁷GpppN where N is any ribonucleotide. The triphosphate bridge can be cleaved by the decapping enzyme DCP2, which hydrolyses the α-β phosphoanhydride bond and DXO, which has both pyrophosphatase and 5'-3' exonuclease activity. The first nucleotide also features a free 2'-hydroxyl in the absence of 2'-O-methylation that can form hydrogen bonds with cytosolic pattern-recognition receptors, most notably IFIT1, a protein which binds the cap in competition with eIF4E, and induces translational arrest. The ribose of the first nucleotide is thus important: if the 2'-hydroxyl is free the RNA is marked as foreign and downstream activation of RNase L and oligoadenylate synthetase can lead to transcript degradation and the production of interferon-inducing oligonucleotides. Cap 0 is typically generated during in-vitro transcription by the inclusion of a simple guanosine cap analog but this method is inefficient, since the analog can be added in either a forward or reverse orientation; the reverse form does not recruit the translation initiation factors and instead acts as a decoy that reduces overall protein yield.
Cap 1 is formed when a methyl group is added to the 2'-hydroxyl position of the ribose of the first nucleotide, producing the structure m⁷GpppNm (where the superscript m indicates the 2'-O-methyl modification). This single methyl addition sterically hinders binding of IFIT1 by occupying a hydrogen-bond donor site the protein's cap-binding pocket employs to distinguish self and non-self RNA. The methylation is catalyzed post-transcriptionally by the 2'-O-methyltransferase enzyme which uses S-adenosyl-L-methionine as the methyl donor and recognizes the N7-methylguanosine cap as the substrate anchor. Synthetically, Cap 1 can be installed co-transcriptionally using trinucleotide cap analogs, such as CleanCap AG, that have the 2'-O-methylated guanosine as the first transcribed nucleotide. This results in a fully methylated transcript that is properly oriented, without the need of a separate enzymatic step. The 2'-O-methyl group also confers improved resistance to the decapping enzyme DXO, but the effect is weaker than that of Cap 2.
Cap 2 carries the 2'-O-methylation pattern to the second nucleotide, which results in the structure m⁷GpppN(m)(p)N(m)(p) where both of the first and second ribonucleotides are methylated on their 2'-hydroxyls. This second methylation is carried out by a separate methyltransferase, Cap 2'-O-ribose methyltransferase, which uses the Cap 1 structure as a substrate and transfers a methyl group from S-adenosyl-L-methionine to the 2'-position of the second nucleotide. This reaction is less efficient than the previous methylation, so Cap 2 is usually only a minor species in most mammalian transcripts, but its functional impact is amplified under conditions of interferon stress where DXO activity is increased. DXO has the capacity to degrade both Cap 0 and Cap 1 transcripts by hydrolyzing the triphosphate bridge and then chewing back the 5' sequence; the second 2'-O-methyl group causes steric hindrance of DXO binding, so it has a protective effect that can increase mRNA half-life by several-fold in cells that are treated with interferon-γ or other inflammatory cytokines. From a synthetic perspective, Cap 2 can be produced using tetranucleotide cap analogs, but these are both more expensive and less efficiently incorporated than the trinucleotide analogs.
The gradual addition of 2'-O-methyl groups establishes a functional gradient with a direct bearing on translation efficiency, immune evasion and decapping sensitivity. Cap 0 is identified as non-self by IFIT1, which outcompetes eIF4E for cap binding, causing translation arrest and RNase L-mediated mRNA decay, interferon induction and low protein production. Steric hindrance of IFIT1 binding by the single methyl group in Cap 1 restores eIF4E recruitment, markedly boosting translation efficiency while dampening interferon activation by several orders of magnitude. Cap 1 also increases resistance to DCP2-mediated decapping, albeit more weakly than the additional methyl group in Cap 2. The second methylation in Cap 2 additionally guards against DXO, an interferon-inducible pyrophosphatase/5'-3' exonuclease that shortens mRNA half-life in an inflammatory milieu. Cap 2 also decreases recognition by RIG-I, a cytosolic helicase that binds to unmethylated 5' ends and induces antiviral signalling, constituting an additional layer of immune evasion that is especially useful when the mRNA is targeted to professional antigen-presenting cells.
Cap 0 to Cap 1 to Cap 2 is a continuum of progressively reduced innate immune stimulation and progressively increased translational efficiency and nuclease resistance. As a result, there is a continuum of properties, which, depending on the desired use, may or may not be favorable. Cap 0 induces interferon and is poorly translated, for example, making it well suited for used for an adjuvanted cancer vaccine. Cap 1 does not activate IFIT1 and can provide high levels of expression without inflammation, and is thus generally the default choice for most vaccines. Cap 2 is resistant to DXO, which can provide greater half-life in interferon-α rich microenvironments, and can therefore be chosen for applications such as gene editing where longer term protein expression is required.
Cap structure also directly controls 5'→3' exoribonuclease susceptibility, and the removal of capping by cytoplasmic decapping enzymes is an important mechanism of uncapped and poorly capped transcript degradation. Cap 0 confers modest XRN1 resistance, but transcripts are still cleaved by the DXO enzyme, which has a specificity for unmethylated first-nucleotide ends and shortens their functional half-life. Masking of the ribose 2'-OH in Cap 1 decreases DXO affinity and allows stability in the hours-to-days range, and Cap 2 additionally impairs the nuclear exosome and NUDT16 decapping factor recognition, making such transcripts last for weeks in non-dividing cells. The triphosphate bridge itself is also susceptible to cleavage; methylation of N7 sterically obstructs phosphatases, and the two additional 2'-O-methyl groups decrease the flexibility of the RNA backbone and make it a less reactive target for metal-catalyzed hydrolysis. Capped mRNAs also have a lower renal filtration rate in systemic circulation because their compact structure decreases hydrodynamic radius and their net charge (which is near zero at physiological pH) does not repel them from glomerular basement membranes. Lyophilized formulations are also more stable when capped due to the cap-dependent increase in glass-transition temperature, with Cap 1 and Cap 2 requiring less lyoprotectant to remain amorphous when freeze-dried.
Translation initiation factor eIF4E recognizes the N7-methylguanosine moiety, regardless of cap type. However, binding is stronger with each additional methylation due to increased hydrophobic contacts and decreased electrostatic repulsion. Transcripts with Cap 1 and Cap 2 are loaded with ribosomes more efficiently than those with Cap 0 because the 2'-O-methyl groups relieve a steric clash between the cap-binding pocket and the ribose backbone, allowing more effective scanning of the 5'-UTR. The cap also supports circularisation through eIF4G–PABP bridging, with Cap 1 and Cap 2 mRNAs forming closed-loop complexes more readily than Cap 0, leading to enhanced ribosomal recycling and more proteins produced per mRNA. in vitro translation of Cap 1 mRNAs results in three- to five-fold more protein than Cap 0, with Cap 2 conferring a small additional increase. The cap also affects start-codon fidelity: transcripts lacking a methyl group often undergo leaky scanning and initiation at downstream AUG codons.
Cap 0 transcripts are sensed by RIG-I and TLR7/8 as foreign RNA. The unmethylated ribose 2'-OH in the cap is structurally similar to viral genomes. Engagement of RIG-I and TLR7/8 leads to activation of IRF3 and NF-κB, transcription factors which induce type-I interferons and pro-inflammatory cytokines. This leads to activation of PKR, which phosphorylates eIF2α, leading to global translational shutdown, and OAS, which generates 2'-5' oligoadenylates which bind and activate RNase L. RNase L degrades both the therapeutic transcript and host mRNAs. Cap 1 abrogates recognition by IFIT1 and IFIT5, cytosolic proteins which bind to the unmethylated cap and block translation initiation, thus decreasing interferon induction by > 90 %. The 2'-O-methyl group also inhibits engagement of TLR7/8 in endosomes, resulting in decreased activation of dendritic cells and less reactogenicity if the mRNA is used as a vaccine. Cap 2 provides further protection against the OAS/RNase L pathway by masking the second nucleotide, which is a preferred substrate for OAS activation, which is particularly important in chronic-dosing regimens where repeated interferon induction could lead to tachyphylaxis or autoimmunity. The cap structure also affects phagocyte uptake, with macrophages preferentially ingesting Cap 0 mRNA via scavenger receptors, while Cap 1 and Cap 2 evade this clearance pathway and thus have prolonged circulation, which can enhance delivery to target tissues.
For prophylactic vaccines, transient expression and a mild innate activation are tolerable: Cap 0 or Cap 1 will suffice, since the therapeutic need is for rapid antigen expression to elicit adaptive immunity. In contrast, for therapeutic protein-replacement (e.g. erythropoietin, factor IX or enzyme-replacement in lysosomal storage diseases), long-term expression with minimal innate inflammation is key, so Cap 1 is the default and Cap 2 the emerging standard for once-monthly or -quarterly dosing. In the context of genome-editing, massive interferon signaling can lead to off-target DNA breaks or cell death; Cap 1 or Cap 2 is necessary to keep cells alive and editing. For cancer vaccines or neo-antigen therapies, some degree of innate activation might be desirable as an adjuvant, so Cap 0 may be leveraged intentionally to heighten immunogenicity. On saRNA platforms, the replicon is highly immunostimulatory, so reducing this load using Cap 2 can prolong the replication window and thus produce more protein, without requiring a higher dose. The cap structure is also a regulatory determinant: Capped with Cap 1 or Cap 2, the transcript is more likely to be approved for chronic administration, as its diminished immunogenicity will fall below safety thresholds for repeat dosing. For low-dose therapies against rare diseases, Cap 1 will ensure that every molecule is translationally competent, maximizing therapeutic impact and reducing cost-of-goods.
In general, Cap 0, Cap 1, and Cap 2 analogs occupy different therapeutic spaces which are demarcated by a tradeoff between desired immunogenicity, needed duration of protein expression, and scalability of manufacturing. Cap 0 is intentionally immunogenic and is used in research, and as an adjuvant for cancer immunotherapy, to elicit interferon pathways. Cap 1 has become a near-universal standard for prophylactic vaccines and metabolic disease therapies, as it is both IFIT1-arrest-resistant, has very high capping efficiency, and is cost-effective. Cap 2 is used for specialized therapeutic applications such as gene editing, oncology, and long-term protein replacement where extended expression is needed in the presence of interferon response or other factors inducing interferon micro-environments. Selection of Cap 1 versus Cap 2 is further nuanced by cell-line and species specific expression patterns of these innate immune sensors.
Cap 0 analogs are the workhorse of basic mRNAs research. Due to low degree of methylation, the immune recognition of such analogs is maintained allowing for the research and development of the RIG-I and TLR7/8 cascades. This is possible due to a predictable immunogenicity of such compounds for mechanistic study. For example, interferon induction, dendritic cell maturation, or adjuvant activity can be evaluated for thousands of candidate compounds at a time. Cap 0 analogs are more suitable for early development stages of vaccines that wish to leave some inflammatory tone in their mRNA compared to Cap 1 analogs. In addition, the lack of 2' O-methyl modifications makes Cap 0 production cheaper and less complex. The reduced synthesis steps limit the chance for aggregation or other undesired secondary events. These make Cap 0 an attractive option for large-scale screenings in which the yield of capping reactions is of lower priority compared to the immune response.
Cap 1 mimics are now the clinical standard for prophylactic vaccines and protein-replacement therapeutics. The 2'-O-methyl group on the first nucleotide attenuates RIG-I and TLR7/8 signalling while maintaining good eIF4E binding and ribosomal recycling. This has the dual effect of allowing high levels of antigen production to prime the adaptive immune response, but without inducing a high level of interferon production which would suppress this response. For protein-replacement therapeutics, Cap 1 mRNAs encoding erythropoietin, clotting factors or metabolic enzymes allow translation to continue for a period of several days, matching the pharmacodynamic window. The cap also contributes to synergy with the poly(A)-tail, increasing circularisation and ribosomal re-initiation to improve the amount of protein product per transcript and decrease the effective dose of mRNA required. Scalable manufacturing has been well developed, and GMP-grade trinucleotide Cap 1 analogs have mature toxicological packages which meet the requirements of regulators.
Cap 2 analogs have 2'-O-methylation present on both the first and second nucleotide. These caps are currently only used in very specific scenarios, where immune evasion and prolonged expression is absolutely required. The additional 2'O-methylation allows for even further resistance to the immune sensors OAS and RNase L. Cap 2 is thus the cap of choice in settings where chronic-dosing is required to maintain expression or therapeutic effect, such as once-monthly enzyme-replacement or long-acting protein therapies, where low-grade activation of interferon cannot be tolerated over time. Cap 2 is also advantageous in neo-antigen cancer vaccines, where minimization of background inflammation that could potentially mask tumor-specific activation is critical to be able to clearly and specifically monitor for a vaccine response. The prolonged half-life is also advantageous in the context of self-amplifying RNA platforms, where long-term persistence of the replicon without negative feedback is required for high-level expression. The additional synthesis complexity of Cap 2 (via synthesis of tetranucleotide level analogs or enzymatic stepwise addition of the methyl group) is outweighed by the improvement in translation in these specific clinical scenarios.
Choice of Cap is dependent on host cell type and species. TLR7/8 and RIG-I, which can recognize Cap 0, are highly expressed in human monocyte-derived dendritic cells (DCs) and monocytes. This limits the choice of Cap for these cell types to Cap 1 or Cap 2 to prevent hyperstimulation of cytokine secretion leading to host cell death. On the other hand, mouse cells are less sensitive to Cap 0 because their TLR profile differs from human cells. Cap 0 is thus viable for preclinical vaccines where stimulation of an immune response is desirable. Insect cells are lacking all proteins used by mammalian cells to detect cap structures and are therefore insensitive to cap methylation. On the other hand, primary human T cells and hepatocytes are sensitive to Cap 0 and Cap 1, and require Cap 1 to avoid toxicities and achieve high expression levels. Finally, for xenogeneic in vivo delivery of human mRNA in mouse, Cap 2 is preferred to prevent detection of the transcript by the murine immune system which would interfere with the readout of translation. RIG-I and TLRs in non-human primates (NHPs) are homologous to human receptors and these cells have similar responses to changes in mRNA cap.
Choice between Cap 0, Cap 1 or Cap 2 analogs is determined by a 4-way tradeoff between desired immunogenicity, necessary duration of expression, complexity of manufacture, and burden of regulatory requirements. Cap 0 is restricted to preclinical and immune-modulating applications in oncology, Cap 1 is the default choice for most vaccines and proteins targeting metabolic disease, and Cap 2 is the premium choice for applications like gene editing or chronic therapies where the extra costs and qualification effort of the longer-lasting product are easily justified by maximal duration of persistence. No choice is best for all indications; the best choice depends on the therapeutic window of the target indication, the interferon status of the target tissue, the desired dose frequency, and the desire to incorporate additional synthetic steps and analytical characterization.
Research-grade cap analogs are manufactured using synthetic routes that are less expensive and faster, but do not emphasize purity. The resulting materials are contaminated with a diastereomeric mixture, residual solvents, and trace heavy metals that are permissible for use in animal studies, but would not be acceptable for use in human studies. Cap 0 and simple dinucleotide caps can be synthesized using one-step phosphorylation chemistry with a yield greater than 80%, which makes these reagents affordable for HTS campaigns and proof-of-concept studies where only low doses in small rodents are needed. Clinical-grade caps, on the other hand, must be produced in GMP facilities with specifications for diastereomeric purity, endotoxin, and mutagenic impurities limits that meet ICH M7 criteria; for Cap 1 and Cap 2, this involves chiral chromatographic resolution to purify the desired diastereomer, which results in a three-fold increase in the manufacturing cost but is required to prevent off-target effects. The difference also applies to the analytical burden: while research-grade materials need only be qualified by HPLC purity, clinical-grade caps require NMR characterization, mass spectrometry confirmation of the molecular weight, and accelerated stability data.
Cap analogs themselves are of course important starting materials, with material composition directly relevant to safety and efficacy, and as such are regulated as such and require full CMC data, including synthetic route, expected impurities, and stability data. Cap 0 is usually considered a well-understood starting material, though its immunogenicity, and the interferon response it causes, is not acceptable for use in prophylactic vaccines; it is sometimes used for adjuvanted cancer immunotherapies where such inflammation is part of the desired effect. Cap 1 is considered the industry standard, with existing acceptance criteria (capping efficiency >80%, residual uncapped<5%, and the cap analog itself being made from starting materials which are either compendial or which have a qualified package of toxicology data). Cap 2 is considered a novel chemical entity on its own, due to the additional chiral center introduced by the second 2'-O-methylation and the need for an additional methyltransferase. As such, developers are expected to provide data showing that the additional enzyme does not also perform off-target methylation elsewhere in the transcript, that the final product is less than 1% of the incorrect diastereomer, safety pharmacology data showing that Cap 2 does not modulate the immunogenicity of the protein it encodes, and extended repeated-dose toxicology in a non-human primate needs to be increased to ninety days to preclude cumulative effects. Regulatory agencies also generally require second-vendor qualification if a single source is used for the cap analog, due to risk of supply disruptions, adding another six to twelve months to development.
Cap selection is a direct trade-off between the marginal cost of added methylation and the performance gain to stability/expression. Economic models indicate that Cap 1 has a fifteen to twenty percent premium on raw-material cost over Cap 0, while Cap 2 requires twice as much starting material due to lower synthetic yield and the requirement for chiral resolution. On vaccines where billion-plus dosing is projected, the premium is easily amortised by the five- to ten-fold translation efficiency gain, and the abrogation of interferon-mediated reactogenicity that reduces effective dose per patient and the cost of adverse-event monitoring. For metabolic disease therapies, where mRNA is delivered weekly, Cap 2 half-life extension can enable twice-weekly dosing regimens, improving compliance and reducing the total cost-of-treatment, despite higher per-dose material costs. In gene-editing, resistance to DXO can improve editing efficiency by up to thirty percent with Cap 2, reducing mRNA dose and the amount of lipid needed for delivery, with an associated potential reduction in chronic toxicology. Performance can also be improved by choosing the first nucleotide: N6-methyladenosine incorporation at the +1 position acts synergistically with Cap 1 to enhance translation, providing a less expensive alternative to Cap 2 for use cases with a more moderate requirement for extended expression. The caveat here is the added regulatory burden: co-transcriptional installation of m6A means the extent of modification must be validated to prove full m6A was installed.
The scalability is dependent on the availability of metric-tonne-scale production of the cap analog in diastereomeric and endotoxin purity that is consistent between batches. The synthesis of Cap 0 involves simple phosphorylation chemistry that is easily scalable in a typical nucleoside production facility, with several validated suppliers creating a more secure supply network. Cap 1 requires resolution of the 2'-O-methylated diastereomer, either by enzymatic methylation with recombinant 2'-O-methyltransferase or chemical synthesis followed by preparative chiral chromatography, both of which have been scaled to the hundred-kg level, but have fewer suppliers, thus creating a bottleneck and an allocation shortage that has been encountered during a pandemic-scale vaccine distribution. Cap 2 is similarly limited in that the second methylation step incurs a thirty percent loss in the overall yield and the final purification of the desired diastereomer requires two tandem chiral columns, a process that is only performed at a few GMP compliant facilities. Developers are attempting to overcome this risk by creating a one-pot enzymatic cascade where a single, engineered methyltransferase installs both 2'-O-methylations, potentially doubling the yield and simplifying the downstream purification. An alternate approach would be to produce a mixed-cap material of ninety percent Cap 1 and ten percent Cap 2 that would have similar stability as Cap 2 without the additional synthetic burden. Regulators have suggested that they would be amenable to such a strategy if the ratio could be tightly controlled and the pharmacology and performance of the mixed-cap molecule could be shown equivalent to that of pure Cap 2 in NHPs. Scalability in the long-term would require that the cap synthesis be distributed beyond single-source supply.
Table 2 Decision matrix for cap selection in therapeutic development.
| Selection criterion | Cap 0 | Cap 1 | Cap 2 | Mitigation strategy |
| Immune activation | High, triggers IFIT1 | Minimal, stealth | Minimal, stealth | Use Cap 0 only for oncology |
| Expression duration | Hours | 1–2 days | 3–5 days | Match to dosing interval |
| Manufacturing cost | Low (+0 %) | Moderate (+15–20 %) | High (+100 %) | Enzymatic methylation |
| Regulatory pathway | Well-understood | Standard IND | Extended tox | Blend Cap 1/Cap 2 |
| Supply security | Multi-vendor | Few vendors | Very few vendors | Second-source qualification |
We offer a comprehensive portfolio of high-quality cap analogs designed to support the full spectrum of mRNA synthesis applications, from early-stage research to commercial production. Our product range includes Cap 0, Cap 1, and Cap 2 analogs, manufactured under stringent quality control standards to ensure consistent performance in in vitro transcription (IVT) workflows.
We provide a full range of high-purity Cap 0 and Cap 1 analogs designed for reliable performance in in vitro transcription (IVT) mRNA synthesis. Our Cap 0 analogs are well suited for basic research, screening studies, and early-stage development, offering efficient incorporation during IVT and robust translation performance. For applications requiring improved biological relevance, our Cap 1 analogs include a 2'-O-methyl modification that more closely mimics native eukaryotic mRNA, supporting enhanced translation efficiency and reduced innate immune activation. All Cap 0 and Cap 1 analogs are manufactured using tightly controlled synthetic processes to achieve high chemical purity and low impurity levels, ensuring consistent results across a wide range of IVT systems.
For commercial applications, we offer GMP-grade cap analogs specifically developed to meet the stringent requirements of therapeutic mRNA production. These products are manufactured under GMP-compliant conditions with comprehensive quality documentation, making them suitable for use in mRNA vaccines, gene therapy programs, and other regulated applications. Our GMP-grade cap analogs are designed to support scalable manufacturing while maintaining critical quality attributes, including structural integrity, purity, and compatibility with validated IVT workflows.
Consistent capping performance depends on reliable raw material quality. Our cap analogs are subject to strict quality control at every stage of production, with thorough analytical characterization to ensure batch-to-batch reproducibility. Key parameters such as purity, impurity profile, and chemical identity are carefully monitored to minimize variability and support reproducible mRNA synthesis results. This level of consistency is especially important for process development, scale-up, and long-term manufacturing, where material variability can directly impact mRNA yield and translation efficiency.
In addition to our standard product portfolio, we offer custom cap analog development to address specialized research or manufacturing needs. Custom options include alternative cap structures, tailored purity specifications, and formulation adjustments to align with specific IVT conditions or process requirements. Our technical team works closely with customers to design and supply custom cap analogs that integrate seamlessly into existing workflows, supporting both exploratory research and large-scale mRNA production programs.
Choosing the appropriate cap analog is a key step in achieving optimal mRNA stability, translation efficiency, and product consistency, and the best option depends on your application, development stage, and regulatory goals. Whether you require high-purity research-grade materials, GMP-grade cap analogs for therapeutic mRNA, or custom cap structures for specialized workflows, our team is ready to support your project—contact us today to discuss your requirements and request a technical consultation or quotation.
References
Cap 0 contains a methylated guanosine cap, Cap 1 adds a 2'-O-methyl group on the first nucleotide, and Cap 2 includes additional methylation on the second nucleotide.
Cap 1 and Cap 2 more closely resemble native eukaryotic mRNA, supporting improved translation efficiency and reduced immune recognition.
Cap 0 analogs are commonly used in basic research and early-stage mRNA studies where standard translation performance is sufficient.
Cap 2 analogs are used less frequently and are typically reserved for specialized applications requiring advanced cap structures.
Yes, additional methylation in Cap 1 and Cap 2 can improve mRNA stability and translation performance.


 Loading ......
Loading ......