The decision to use Cap 0 or Cap 1 analogs is a strategic choice that weighs immunogenicity, translational efficiency, and manufacturing complexity against the therapeutic goal. Cap 1 is the clear choice for clinical applications where high protein expression and low innate immune activation are crucial, as its 2'-O-methylation imparts immune evasion and a longer half-life. Cap 0 only has a place in experimental settings where interferon induction is intentionally used as an adjuvant or where cost considerations may be prioritized over performance. The difference in structure — the absence or presence of a single methyl group — leads to a functional gap that determines whether the transcript will be recognized as foreign or self, directly affecting translation rates and safety profiles.
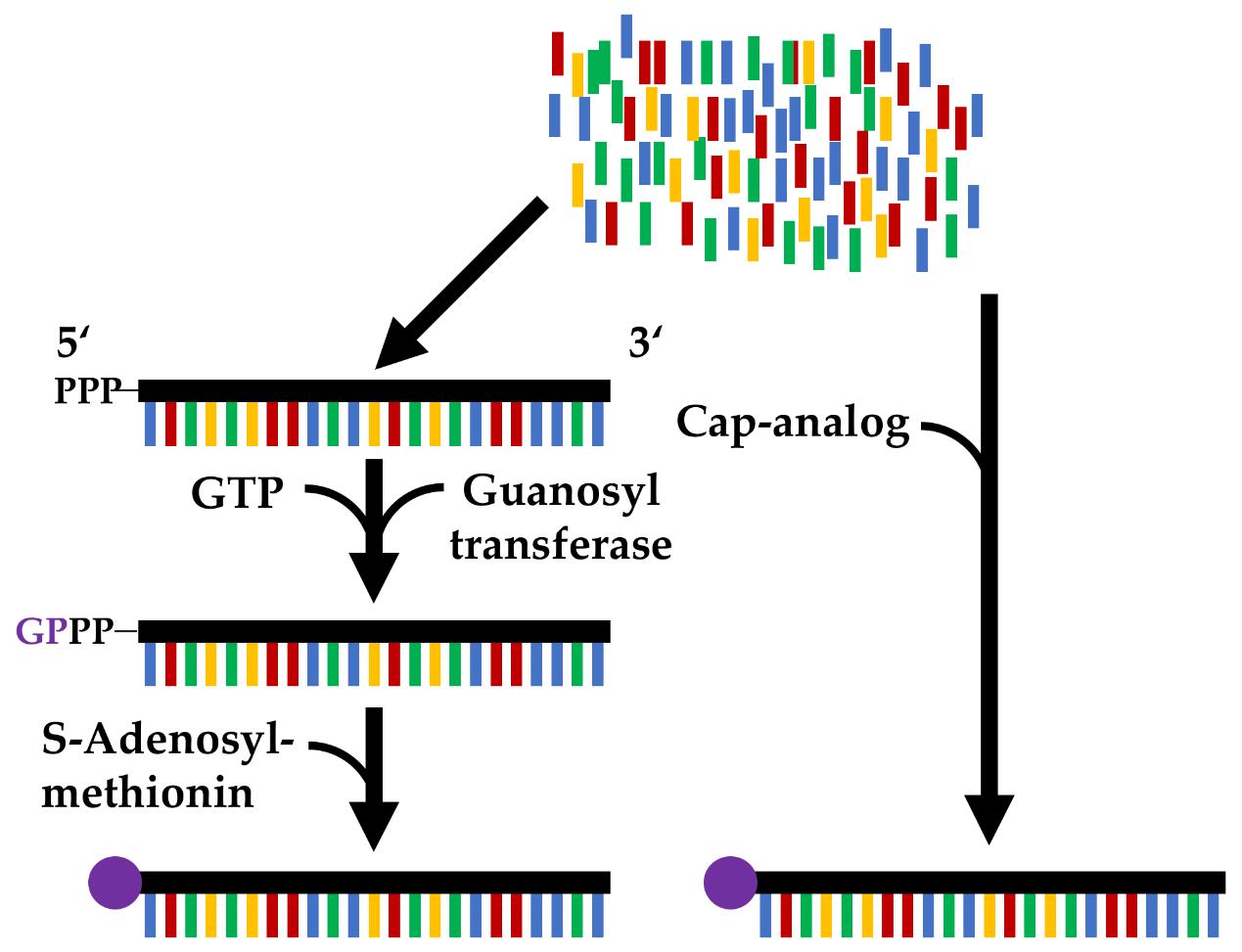 Fig. 1 Comparison of the post-transcriptional capping (left) with a two-step enzymatic reaction and the co-transcriptional capping (right) by adding a cap analog, catalyzed by the enzyme used in the in vitro transcription.1,5
Fig. 1 Comparison of the post-transcriptional capping (left) with a two-step enzymatic reaction and the co-transcriptional capping (right) by adding a cap analog, catalyzed by the enzyme used in the in vitro transcription.1,5
Cap 0 and Cap 1 are two levels of the 5' terminal methylation that underlie differences in performance of synthetic mRNAs. They both have the same basic framework: a 7-methylguanosine group connected to the first transcribed nucleotide by an inverted 5',5'-triphosphate bridge. This structure allows for minimal recognition by translation initiation factors and limited resistance to 5'-to-3' exonucleases. The key difference is whether or not the ribose on the first transcribed nucleotide is methylated. In the Cap 0 structure, the cap ends here, and the penultimate nucleotide remains unmodified except for its normal ribose hydroxyl groups. In the Cap 1 structure, an enzyme transfers a methyl group to the 2'-O of the first transcribed nucleotide, resulting in a more complex structure. The presence of this modification is very important for recognition, as the cell will see the Cap 1 structure as "self" RNA and the Cap 0 structure as potentially foreign. The structural difference also affects translation initiation rates, sensitivity to decapping enzymes, and activation of innate immune sensors. This results in different stability and expression profiles, making the choice between the two important for optimizing mRNA therapeutics.
The primary difference between Cap 0 and Cap 1 is the methyl group at the first transcribed nucleotide. The differential methylation has implications in how the translation and immune systems recognize the Cap. Cap 0 is solely an N7-methylguanosine cap linked to a nucleoside (almost always guanosine) by a 5'–5' triphosphate bridge. It is the most basic form of cap that can be biologically active, and allows for basic exonuclease protection and binding of eIF4E. Cap 1 is N7-methylguanosine cap linked to a 2'-O-methylated nucleoside. The additional methyl group at the 2' hydroxyl of the ribose in the first nucleotide creates a dimethylated nucleoside. This results in a chemical structure that has a statistical probability of being identical to the cap present on any mammalian mRNA. This additional methylation causes steric clashes which cause lower binding to pattern recognition receptors and higher affinity for eIF4E due to an optimized van der Waals force. The phosphate backbone may also be altered as newer Cap 1 analogues have used longer polyphosphates and phosphorothioates for increased stability from enzymatic cleavage.
Distribution of cap 0 and cap 1 structures is consistent with their historical development as a reflection of the immune pressures found in the host cell. Cap 0 is the original cap structure, and is most common in unicellular eukaryotes (yeast, plants, invertebrates), where complex innate immune surveillance may be lacking or not as stringent. In many viruses, 2'-O methylation is lacking (due to lack of methyltransferase enzymes) which is why mRNA with a cap 0 structure is able to induce a strong interferon response when transfected into mammalian cells. On the other hand, cap 1 is the predominant form in vertebrates, in particular mammals. Here the methyltransferase activity is constitutively active and coupled to transcription. The evolutionary expansion of complex pattern recognition machinery to distinguish between self and non-self RNA would be expected to select for cap 1, as the 2'-O methyl group can be used as a signal to bypass the interferon-inducible proteins and other recognition proteins that specifically bind to unmethylated cap 0 structures. This should also be considered when selecting a cap analog for a specific application (e.g., for expression in mammalian cells, cap 0 has characteristics of a viral infection while cap 1 is undetectable by the immune system).
Table 1 The features of Caps.
| Feature | Cap 0 | Cap 1 | Evolutionary/Biological Significance |
| Methylation | N7-methylguanosine only | Additional 2'-O-methyl on first nucleotide | Immune evasion mechanism |
| Natural Hosts | Yeast, fungi, protists | Mammals, higher vertebrates | Complexity correlation |
| Immune Response | Triggers innate activation | Recognized as self RNA | Self vs. non-self discrimination |
| Translation Factor Binding | Moderate eIF4E affinity | Enhanced binding affinity | Efficiency optimization |
| Processing Pathways | Basic splicing/export | Coordinated maturation | Quality control integration |
The differences between Cap 0 and Cap 1 come down to a single methyl group. This group changes the identity of the 5' end of an RNA molecule, from a sign of danger (pathogen-associated molecular pattern) to a sign of self (self-signature) that is not recognized by the host immune system. In doing so, it epigenetically gates translation initiation, innate immune escape and transcript stability so that Cap 1 takes primacy over any use that requires ongoing protein expression in a mammalian system. Cap 0 has the minimum N7-methylguanosine cap that is required for eIF4E recruitment, but the transcript remains IFIT1-bound, unable to scan for a start codon and triggering an interferon response leading to transcript decay and inflammatory response. The 2'-O-methyl group on Cap 1 sterically hinders this binding, allowing continued translation and a lower susceptibility to interferon-inducible decapping enzyme DXO, important in cells that have high expression of inflammatory cytokines. This results in different stabilities: Cap 0 mRNAs have half-lives in the order of hours under normal conditions, whereas Cap 1 mRNAs can last days, allowing for less frequent administration and more predictable pharmacokinetics. These differences are not just quantitative but also qualitative, in that they decide if a transcript is going to be translated or not, if it is going to be destroyed or not, if it is going to be ignored by the immune system or not. The difference also manifests in manufacturing costs, as Cap 1 synthesis requires more steps than Cap 0, but is necessary to meet safety and efficacy profiles expected in human systems.
In terms of translation efficiency, Cap 1 is superior to Cap 0. This is due to an overall higher level of protein expression attributed to bypassing IFIT1-mediated translational arrest and subsequent 43S pre-initiation complex (PIC) stalling by permitting ribosomal scanning to reach completion and encounter the true start codon. Both cap structures are bound by the cap-binding protein eIF4E through the N7-methylguanosine moiety with the same affinity, but the methylated 2'-hydroxyl group of Cap 1 sterically hinders IFIT1 binding and permits eIF4E to form eIF4F complex, unwind the 5' UTR and license scanning that ultimately results in start codon recognition and elongation. As a result, relative to Cap 0, Cap 1 has a 5- to 10-fold higher expression of a luciferase reporter in primary human dendritic cells as measured by either immunoblot or flow cytometry, which is also due to increased transcript stability. This is especially true for professional antigen-presenting cells like dendritic cells, which constitutively express IFIT1, where at 72 hours Cap 1 mRNA transcripts are still highly expressed and Cap 0 mRNA is no longer detected 12 hours post-transfection. The extended half-life of Cap 1 in this context also further enhances translation by allowing more ribosomal loading events per transcript to take place. Consequently, it is more efficient to utilize Cap 1 mRNA for protein production, since higher protein expression can be reached at lower doses compared to Cap 0. The cost-of-goods is lower due to less input required, and there is less risk of off-target effects that are often associated with excess mRNA accumulation in the cytosol.
The human innate immune system can distinguish Cap 0 from Cap 1 with high sensitivity. The lack of 2'-O-methylation on the ribose moiety in Cap 0 is an innate immune system PAMP that activates strong interferon production. IFIT1, for example, directly recognizes the unmethylated ribose of Cap 0 and antagonizes eIF4E binding, triggering a potent interferon response. These interferons can induce a global antiviral state (including translational arrest, RNase L activation, and production of more type I IFNs) and the transcriptional induction of other restriction factors that create a toxic environment for any remaining transcripts. This can reduce bystander mRNA expression and result in off-target inflammation that would be unacceptable in prophylactic vaccination. RIG-I and MDA5 (sensing of uncapped or improperly capped RNA) have increased affinity for Cap 0 transcripts, especially if the 5' terminus is incompletely methylated, and initiate MAVS-dependent induction of more interferon. Cap 1 prevents these responses by sterically blocking IFIT1 binding due to its methylated ribose and showing reduced binding to RIG-I, making the transcript invisible to the immune system. Cap 0's immunogenicity may be useful in cancer treatment where you may want to activate interferon signalling in the tumor micro-environment to enhance cross-presentation of tumor antigens and induce anti-tumor immunity.
Cap 1 forms a considerably more stable and longer-lived cap structure than Cap 0 by several different, synergistic mechanisms. The added 2'-O-methyl group on the first nucleotide inhibits the access of decapping enzymes (such as Dcp1/Dcp2) that would otherwise remove the cap to flag the transcript for 5'–3' degradation, due to steric hindrance. Cap 1 is also a stronger eIF4E binder, which allows it to protect itself from decapping by competitive inhibition. Cap 1 is also more resistant to 5'–3' exonucleases due to the extra methylation group that increases the steric hindrance of the inverted 5'–5' triphosphate linkage. Cap 1 transcripts are also less prone to being trafficked to processing bodies where mRNA degradation machinery is often found in cells, and more likely to remain in the translating pool, where ribosomes can shield it from degradation. These effects make Cap 1 significantly more stable in vivo and in primary cells than Cap 0, where the latter has a half-life of a few hours before being targeted by the immune system while the former remains for days. This allows for a greater total amount of protein to be produced as each transcript can last longer and be used for multiple rounds of translation. In addition to a higher protein yield, it is also less variable and requires a lower dosage between treatment intervals, and requires less mRNA for a given effect so has less side effects and lower manufacturing costs.
Table 2 Functions of Caps.
| Functional Parameter | Cap 0 Characteristics | Cap 1 Characteristics | Clinical Implication |
| Translation Efficiency | Moderate eIF4E binding, reduced initiation | Enhanced eIF4E affinity, efficient initiation | Higher protein expression yields |
| Immune Recognition | Activates RIG-I/MDA5 pathways | Evades pattern recognition receptors | Reduced inflammation, improved safety |
| Decapping Resistance | High Dcp2 hydrolysis rate | Sterically hindered enzyme access | Extended functional half-life |
| Overall Stability | Short persistence in cells | Prolonged intracellular retention | Improved dosing efficiency |
| Therapeutic Applicability | Limited to research contexts | Standard for clinical translation | Regulatory compliance and efficacy |
The choice of cap analog to use in a given application should therefore be based on the intended use. It is a matter of assessing the trade-offs in a variety of important parameters such as immunogenicity, translation efficiency, manufacturability and costs. The requirements for these parameters will be different for the various applications and use cases of mRNA. Experimental flexibility and cost considerations are the most important aspects for research applications, while milder translation and higher reagent and synthesis costs can be tolerated. mRNA vaccines need to find a balance between achieving strong protein expression to elicit an immune response but low enough to avoid activating the innate immune system too much. For mRNA therapeutics, safety and biocompatibility are the main criteria as mRNA is likely to be administered to patients at multiple time points for a possibly long period of time. Cell type, administration route, desired length of protein expression and even the manufacturing process can also affect the choice of cap.
When cost, experimental flexibility, or ease of synthesis are primary concerns, such as for research purposes or high-throughput screening, and reduced translational efficiency is an acceptable trade-off, Cap 0 analogs can be suitable. Cap 0 reagents afford a basic level of nuclease protection and translational activity that is often adequate for research applications. Their simpler synthesis can also lower reagent costs and make them more practical for large-scale experiments where many mRNA sequences need to be tested. Furthermore, in certain organisms like yeast, invertebrates, or plants where Cap 0 is the prevalent cap structure, it is the physiologically relevant choice and does not pose an immunogenicity issue. When using translation systems such as rabbit reticulocyte lysate or wheat germ extract in vitro, where immunogenic response is not a factor, the increased stability of Cap 1 reagents confers little practical benefit. In studies aimed at elucidating the basic principles of cap-dependent translation, Cap 0 may be intentionally used to generate easily removable cap substrates to study the kinetics of cap turnover.
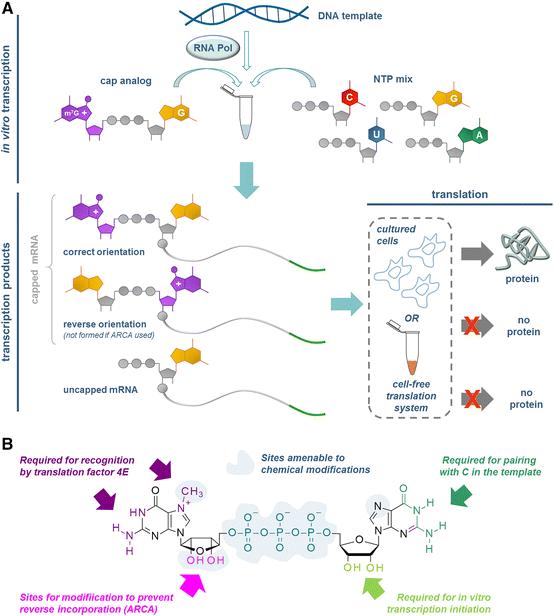 Fig. 2 Synthesis of capped RNAs by so-called co-transcriptional capping during transcription in vitro.2,5
Fig. 2 Synthesis of capped RNAs by so-called co-transcriptional capping during transcription in vitro.2,5
mRNA vaccines require cap analogs that drive the highest protein expression, and have well-controlled innate immune activation in order to have high immunogenicity while still being safe. Cap 1 motifs with further backbone modifications are therefore the best candidates for vaccine development as the 2'-O-methyl modification can mask the transcript as self RNA, thus allowing it to escape recognition by interferon-inducible proteins that would otherwise attenuate antigen production and lead to unwanted inflammatory responses. Escape from innate immune activation is required for mRNA vaccines that are used to fight infectious diseases, in which high, but not prolonged, antigen expression is required in order to elicit an adaptive immune response. The presence of these modifications in the phosphate bridge also play a role in vaccine efficacy; modifications that can block decapping enzymes can extend the expression of the antigen, which can increase immunogenicity while also allowing for a lower dose. The kinetics of capping is also of high importance in vaccine production since uncapped transcripts can be recognized by PRRs, and any uncapped mRNA will lower the fraction of active mRNA in the final product. Trinucleotide capping analogs that directly form Cap 1 can therefore be used for co-transcriptional capping, which simplifies the production process and allows for more uniformity between batches.
Therapeutic applications of mRNA have the most strict requirements on the choice of cap, including safety, long-term expression, and low immunogenicity for chronic or repeated applications. The use of Cap 1 is critical in protein replacement therapies where patients may require lifelong administration. In these cases, any immunogenicity directed towards the mRNA therapeutic could be detrimental to the treatment's efficacy or cause unwanted side effects. The longer half-life of mRNA capped with Cap 1 can decrease the required dosing frequency, which is more convenient for the patient and still maintains therapeutic protein levels. In the context of rare diseases where the treatment options are limited, the improved translational efficiency of Cap 1 ensures maximal protein production from each dose of mRNA, which lowers the cost of manufacture and reduces potential side effects. For cancer immunotherapy where mRNA may encode cytokines or chimeric antigen receptors, Cap 1's lack of immunogenicity is key to repeated dosing without eliciting anti-vector responses that would preclude further administration. Gene editing applications can leverage Cap 1's stability for sustained expression of nucleases or base editors over several days, which can increase editing efficiency while reducing off-target effects. For advanced therapeutic development, Cap 2 structures or cap analogs with longer phosphate linkages are being used for additional stability, particularly for protein expression in slowly dividing cells. In terms of manufacturing, co-transcriptional capping using a trinucleotide analog to directly generate Cap 1 is the most attractive since the resulting homogeneous product can be submitted to regulatory authorities and the method is scalable.
Regulatory and Manufacturing Considerations for Cap Analogs in mRNA Therapeutics: A Comprehensive Guide explores the diverse aspects involved in ensuring the quality, safety, and consistency of cap analogs throughout the drug development and manufacturing process. In mRNA therapeutics manufacturing, control of cap structure, orientation, purity, and immunogenic contaminants is critical, with regulatory agencies demanding detailed documentation of synthetic routes, process validation, and analytical methods to ensure batch-to-batch consistency. Transitioning from lab-scale synthesis to GMP (Good Manufacturing Practice) manufacturing requires the implementation of quality management systems, risk-based approaches, and validated cleaning procedures to prevent cross-contamination. Manufacturers must establish specifications for cap analogs, including chemical identity, stereochemical purity, residual solvents, and demonstrate that synthetic modifications do not introduce toxicological concerns. Manufacturing environment considerations include sterility assurance and environmental monitoring standards, particularly for reagents used in co-transcriptional capping for mRNA drug substance production.
Differences between research grade and GMP cap analogs. The level of quality control, documentation, and regulatory compliance for cap analogs designed for research use compared to those intended for clinical use (GMP) are defined by their end use. Research reagents are made for use in discovery processes with the minimum quality controls, typically identity and purity, and lack the robust validation that is needed for therapeutics. Therefore, small variations between different lots/batches of research cap analogs can be acceptable and will not affect their use in early discovery efforts when proof of concept is the main objective. GMP cap analogs, however, must be made with stringent quality systems in place, detailed process validation, equipment qualification and environmental controls. The facility where GMP cap analogs are manufactured will need to be in compliance with the regulatory guidance for facility and equipment requirements which would also include filing of detailed batch records, certificate of analysis for each manufactured lot, and demonstration of robustness of the manufacturing process with multiple, consecutive production runs before any clinical trial material is released. Validated analytical methods will need to be developed for quality attributes such as cap analogue purity, residual solvents, heavy metals, and microbial contamination.
The scalability and cost factors in the large-scale production of cap analogs are influenced by the synthetic approach, molecular complexity, and required manufacturing capacities. Co-transcriptional capping with dinucleotide or trinucleotide analogs is generally more scalable than post-transcriptional enzymatic capping, as it avoids the need for separate purification steps and minimizes material losses. The cost of synthetic cap analog precursors is a major cost driver in the manufacturing process, with more complex structures requiring multi-step organic synthesis and purification methods that can significantly increase production costs. Extended phosphate linkages and modifications of the backbone, such as phosphorothioates, add to the manufacturing complexity and cost, but these are often offset by improved therapeutic performance. Scaling up production from gram to kilogram quantities involves process intensification, including automation of synthesis and purification processes, in-process controls, and reaction condition optimization for improved yield and quality. Investments in facility and equipment for GMP production, such as dedicated suites with controlled environments, closed-system equipment to prevent contamination, and real-time monitoring systems, contribute to capital expenditures. The cost per dose decreases significantly with increased production scale, making high-throughput manufacturing more economically feasible for commercial products, while materials for early-stage clinical applications remain disproportionately expensive due to small batch sizes and extensive quality testing.
We offer a focused portfolio of Cap 0 and Cap 1 analog solutions designed to support efficient and reproducible mRNA synthesis using in vitro transcription (IVT). Our products are developed to address the distinct performance and application requirements associated with each cap structure, enabling users to select the most appropriate option based on translation efficiency, immune recognition considerations, and workflow complexity.
Our Cap 0 and Cap 1 analogs are manufactured to achieve high chemical purity and low impurity profiles, ensuring reliable incorporation during IVT and consistent performance across a wide range of mRNA applications. Cap 0 analogs are well suited for basic research, screening studies, and early-stage optimization, while Cap 1 analogs provide an additional 2'-O-methyl modification that supports enhanced translation efficiency and improved biological relevance in more advanced mRNA workflows. This range of options allows users to balance performance requirements with process simplicity and cost considerations.
To support regulated mRNA production workflows, we provide GMP supply options for both Cap 0 and Cap 1 analogs, along with comprehensive quality documentation. These materials are manufactured under controlled conditions with rigorous quality control, supporting reproducibility, traceability, and long-term process consistency without compromising performance in IVT systems.
Selecting between Cap 0 and Cap 1 analogs requires a clear understanding of how cap structure influences mRNA stability, translation efficiency, and overall process performance. By aligning cap selection with your specific application and workflow requirements, you can achieve more reliable and reproducible results—contact us today to discuss your mRNA project and request technical guidance or a quotation.
References
Cap 1 includes an additional 2'-O-methyl group that improves biological relevance.
In many systems, Cap 1 supports improved translation, but performance depends on the application.
Yes, Cap 0 is widely used in early-stage and screening studies.
Yes, Cap 1 generally reduces immune recognition compared to Cap 0.
Selection depends on expression requirements, system sensitivity, and workflow goals.


 Loading ......
Loading ......