2'-O-methoxyethyl (2'-MOE) and related sugar modifications have become the structural foundation for next-generation oligonucleotide therapeutics, reshaping first-generation phosphorothioate strands into high-affinity, long-lived drug-like biopolymers. By fixing the ribose in a C3'-endo pucker with a flexible, hydrophilic side chain, MOE simultaneously increases melting temperature, suppresses innate sensors and extends tissue half-life from hours to weeks. The same chemical grammar now underlies approved gapmers, mixmers and siRNA conjugates, validating sugar modification—not backbone chemistry—as the primary lever for potency, safety and commercial scalability.
While first-generation ASO molecules had established proof-of-concept for gene silencing, they were cleared within minutes of administration, triggered cytokine storms and needed doses in the gram range. The field therefore shifted focus to sugar-based edits that could maintain RNase-H recruitment while having drug-like pharmacokinetic profiles. This led to the development of chimeric constructs in which 2'-MOE "wings" flank a DNA gap or in which MOE/OMe mixtures tile the siRNA guide strands. These approaches have resulted in six marketed drugs and multiple others in late-stage development, and have established sugar modification as the standard evolutionary path for systemic RNA therapeutics.
Early phosphorothioate strands suffered rapid renal clearance, dose-limiting complement activation and off-target cleavage due to promiscuous protein binding. Their unmodified ribose also triggered TLR7/8 and RIG-I, producing flu-like symptoms that capped the feasible dose. Finally, low affinity forced the use of 20–25 mer sequences, driving up cost-of-goods and limiting market acceptance. These shortcomings created a clear design brief: boost affinity, mute immunogenicity and extend half-life without sacrificing catalytic mechanisms such as RNase-H or RISC loading.
2'-MOE, 2'-F and 2'-OMe each bias the sugar toward a C3'-endo pucker that deepens the major groove, increases base-stacking enthalpy and raises melting temperature per nucleotide. The same edits remove the nucleophilic 2'-OH, conferring multi-log resistance to serum and tissue RNases while masking the minor-groove pattern recognised by TLR7/8. Because these benefits are achieved with minimal steric penalty, developers can shorten sequences (16–18 mer) yet retain sub-nanomolar affinity, directly translating into lower dose, lower cost and lower toxicity.
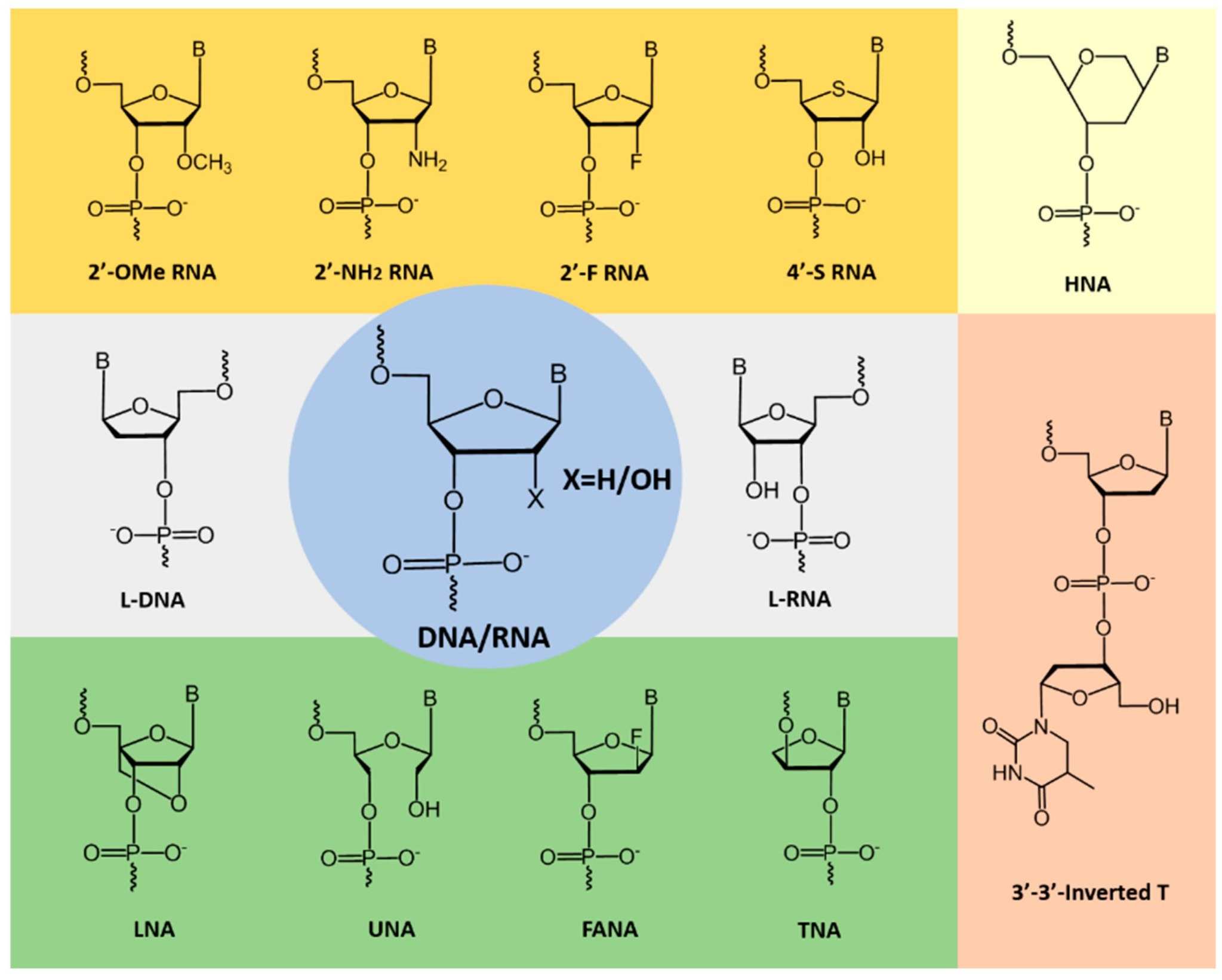 Fig. 1 Examples of chemically-modified nucleotides used to improve aptamer stability and kinetics.1,5
Fig. 1 Examples of chemically-modified nucleotides used to improve aptamer stability and kinetics.1,5
Synthesised for the first time in the mid 1990s, 2'-MOE rapidly emerged as the de facto standard for second-generation ASOs, due to its unparalleled affinity-to-toxicity ratio. The addition of an ethylene-glycol side-chain renders the nucleotide only marginally more lipophilic, just enough to facilitate plasma-protein binding and retard renal filtration, but also remains hydrophilic enough to preclude aggregate formation upon high-concentration formulation. Proof-of-concept clinical programmes in hypercholesterolaemia, spinal muscular atrophy and transthyretin amyloidosis have since validated a quarterly dosing interval, following either subcutaneous or intrathecal administration, entrenching 2'-MOE as the yardstick by which newer sugar analogues are judged.
Table 1 Sugar-Modification Generations
| Generation | Key Chemistry | Primary Gain | Clinical Drawback |
| First | Phosphorothioate backbone | Nuclease resistance | Protein-binding toxicity |
| Second | 2'-MOE, 2'-F, 2'-OMe | Affinity + safety | RNase H loss |
| Third | LNA, cEt, ENA | Ultra-high affinity | Over-stabilization risk |
The 2'-MOE nucleotide is distinguished by the replacement of the ribose 2'-hydroxyl with an O-(2-methoxyethyl) ether. The 2'-MOE group appends a flexible, hydrophilic two-carbon linker terminated with a second ether oxygen, making the molecule sterically bulkier than 2'-OMe while still less lipophilic than the alkyl groups. The extra oxygen functions as a hydrogen-bond acceptor that forms a structured hydration shell around the phosphodiester backbone and the ethylene component provides torsional freedom which prevents the helix from becoming too rigid.
Installations start with transient protection of the 3'- and 5'-hydroxyls (bis-TBDMS or orthoester), generation of the 2'-alkoxide with NaH, and alkylation of 2-bromoethyl methyl ether under strictly anhydrous conditions in THF. The reaction is moisture-sensitive as trace water hydrolyses the activated sugar and yields mixed 2',3'-MOE ethers that co-crystallise and are chromatographically silent. Because the ethylene-glycol arm is flexible, the product often crystallises as a mixed solvate; hence the final protected nucleoside is dried under graduated vacuum to remove bound ether before phosphitylation. The extra synthetic step lengthens the route by one reactor volume and doubles the solvent demand compared to 2'-OMe, but the affinity gain per nucleotide is sufficiently large to allow shorter sequences that offset the cost premium.
In contrast to locked nucleic acids, MOE also has a degree of conformational freedom that may be required for catalytic mechanisms. For gapmer ASOs, flexibility in the central DNA gap is required to permit RNase-H to bind to the heteroduplex, with the MOE wings endowing high local affinity without freezing the entire helix and preventing the enzyme from bending the RNA strand into its active site. In splice-switching mixmers, this flexibility allows the ASO to dislodge spliceosomal proteins while not entrapping the pre-mRNA into a rigid scaffold sterically occluding exon recognition. Molecular-dynamics trajectories further predict that the ethylene-glycol arm samples a wide range of rotamers on the nanosecond timescale, while the most populated state retains the C3'-endo pucker necessary for A-form geometry. This dynamic behavior is conserved in vivo, where the MOE sugar can adapt to other structured RNA elements such as stem-loops or G-quadruplexes without loss of binding energy. Finally, the flexible tether mitigates the risk of over-stabilization that can lead to off-target hybridization, enabling the use of shorter sequences that minimize the potential for seed-region mismatches.
This affinity-flexibility trade-off is modality-specific. In gapmer ASOs, MOE wings are the only modification that can simultaneously hit the sweet spot of high local Tm and the conformational breathing necessary for catalytic turnover. In siRNA, the blended pattern of MOE on the passenger strand and 2'-F on the guide seed harnesses the best of immune silence and ultra-high specificity to enable shorter sequences with lower manufacturing costs and diminished off-target potential. From a regulatory perspective, MOE's decades-long history in chronic dosing studies has established a toxicology precedent that allows for faster new drug applications, while newer constrained sugars must first develop comparable safety databases. Finally, the hydrophilic side chain of MOE reduces the risk of hepatic accumulation that can result from modifications with excessive lipophilicity, which has supported the clean safety profiles observed in multi-year clinical trials for hereditary transthyretin amyloidosis, spinal muscular atrophy and other chronic indications.
2'-MOE is the work-horse sugar of next-generation antisense and siRNA drugs, because a single ethylene-glycol arm simultaneously abolishes the 2'-hydroxyl nucleophile, raises melting temperature, and orders hydrating water in the minor groove. The modification is large enough to sterically hinder RNases, yet flexible enough to preserve the conformational breathing required for RNase-H or RISC catalysis, translating into month-long tissue half-lives and sub-milligram therapeutic doses.
The flexible ethylene-glycol tether adopts multiple rotamers. However, the most populated conformer orients the terminal methoxy into hydrogen-bonding distance of the 3'-phosphate, creating an intramolecular bridge that further rigidifies the sugar. This "soft lock" decreases the conformational entropy of the backbone, and its entropic cost makes it less accessible to the active sites of nucleases, which require a flexible substrate to bind and cleave. Molecular-dynamics simulations have shown that the MOE side-chain orients a shell of hydrating water molecules, which acts as a kinetic barrier, retarding the approach of both serum endonucleases and intracellular RNases. The modification is as effective against 3'-exonucleases that "nibble" termini as it is against endonucleases that cleave internally, enabling the oligonucleotide to transverse extracellular compartments, endosomal vesicles and nuclear speckles without measurable degradation. Finally, the ethylene-glycol arm reduces the risk of over-stabilization, which can lead to off-target hybridization, enabling the use of shorter sequences that minimize the potential for seed-region mismatches.
The enthalpic bonus is synergistic: neighboring MOE sugars lock into the same C3'-endo envelope, establishing a stacking network that further raises the enthalpy of duplex formation. The ethylene-glycol arm polarizes the nucleobase, reinforcing hydrogen-bond donor and acceptor interactions, while leaving the canonical Watson-Crick geometry unchanged. In gapmer ASOs, this MOE wingtip geometry provides high local melting temperature while the central DNA gap remains sufficiently flexible for RNase-H to bind the heteroduplex, a tightrope that no bulkier constrained sugar can walk without over-stabilizing the helix. This higher Tm creates a buffer against single-base mismatches that sharpens therapeutic index and reduces the dose required for maximal reduction of mRNA. Finally, the higher binding energy is not accompanied by a loss of strand invasion efficiency of structured RNA elements such as stem-loops or G-quadruplexes, opening up the possibility of targeting regions of the transcriptome previously considered "undruggable".
The methyl-terminus of the MOE side-chain contributes a small increase in logP which promotes reversible plasma protein binding. Plasma protein binding creates a depot effect that prevents immediate glomerular filtration, prolonging the time that plasma concentrations remain above the efficacy threshold to days following a single sub-cutaneous injection. In tissue, MOE sugar are resistant to exo- and endonucleases, which allows the oligonucleotide to diffuse across the interstitial matrix towards its target cells in the liver, muscle, and CNS compartments. This modification also does not trap the molecule in endosomal compartments. The oligonucleotide is released to the cytosol and nucleus where it can interact with mature mRNA or newly transcribed pre-mRNA transcripts. The widespread biodistribution underpins the clinical utility of MOE-rich gapmers across hepatic, neuromuscular, and ocular indications where durable gene modulation can be accomplished with quarterly or even semi-annual dosing. Finally, the enhanced stability allows for a lower phosphorothioate content, reducing the risk of protein-binding related toxicities such as complement activation or platelet aggregation and allowing for higher cumulative dosing to treat chronic genetic diseases.
2'-MOE nucleotides are the structural basis of most modern antisense drugs, as a single ethylene-glycol arm can increase affinity, hinder nucleases, and mask sensors all at once. With a 2'-MOE flanking each side of a central DNA gap ("winged gapmer"), the resulting duplex has high-affinity and long-life, leading to efficient recruitment of RNase-H for catalytic target degradation, while the 2'-modified sugar itself remains metabolically inactive. This "gapmer" design has led to multiple FDA-approved drugs, and established MOE as the standard sugar for systemic antisense use.
The length of the central DNA gap is usually 8–10 nucleotides. This length is long enough to promote robust RNase-H engagement, while still short enough to prevent undesirable off-target cleavage. MOE wings are typically 4–6 nucleotides in length, which is long enough to impart a high local melting temperature. This keeps the target RNA molecule from dissociating prematurely, before cleavage takes place. The ethylene-glycol arm of MOE also structures hydrating water molecules, which creates a kinetic barrier to exo- and endonuclease attack, prolonging tissue half-life from hours to weeks. MOE itself does not recruit RNase-H; instead, the enzyme's active site is exposed to a flexible RNA: DNA heteroduplex, which can be bent into the catalytic cleft without steric interference. Finally, the gapmer design is amenable to both sub-cutaneous and intrathecal administration, which is necessary for chronic treatment of hepatic, neuromuscular and CNS indications with quarterly, or even semi-annual dosing intervals.
Clinical success is supported by pharmacokinetic data that MOE-rich gapmers distribute to liver, muscle and CNS compartments and remain intact for weeks after a single injection. The hydrophilic side chain of the modification also minimizes non-specific protein binding, diminishing the likelihood of coagulation-factor interactions that bedeviled earlier, fully phosphorothioated versions. Long-term extension studies have demonstrated no cumulative renal or hepatic toxicity at cumulative exposures well above gram-scale doses, a safety margin that has in turn emboldened developers to pursue higher doses and therefore shorter sequence lengths for less accessible tissues like cardiac and skeletal muscle. Finally, the regulatory precedent set by MOE gapmers has simplified chemistry, manufacturing and controls (CMC) packages for follow-on candidates, shortening development timelines and cost.
Contemporary design pipelines balance the thermodynamic stabilization of each MOE substitution, against its predicted effect on RNase-H cleavage rate, off-target seed-match binding stability and liver transaminase levels. The result is usually a pattern which targets MOE to each nucleotide of the wings and leaves the central gap unmodified, providing the highest on-target potency while minimizing the impact on conformational flexibility needed for catalytic turnover. Toxicity is mitigated by avoiding over-methylation of the wings and methylation beyond the wings is usually avoided to prevent excessive-stabilization that would impair RNase-H binding or promote liver sequestration. Finally, the gapmer sequence is screened for repetitive stretches or GC-rich blocks that might cause aggregation or innate immune activation, to ensure the final construct can be manufactured at multi-kilogram scales without chromatographic rescue steps.
In addition to the already existing two categories of ribose modifications (2'-MOE and 2'-fluoro), a third generation of ribose edits that drive affinity, specificity and metabolic stability to even further extremes is emerging. Locked nucleic acids, constrained ethyls, ethylene-bridged analogues, and even cyclohexene scaffolds add extra rings, methyl locks, or ethylene tethers to freeze the furanose ring into a near-perfect A-form helix and to generate new pharmacophoric surfaces for protein recognition. It is hoped that these LNA-like melting increments can be achieved without LNA's concomitant steric rigidity or hepatotoxicity, and thereby permit shorter, more specific sequences that can be dosed at lower total exposure yet remain active for a month.
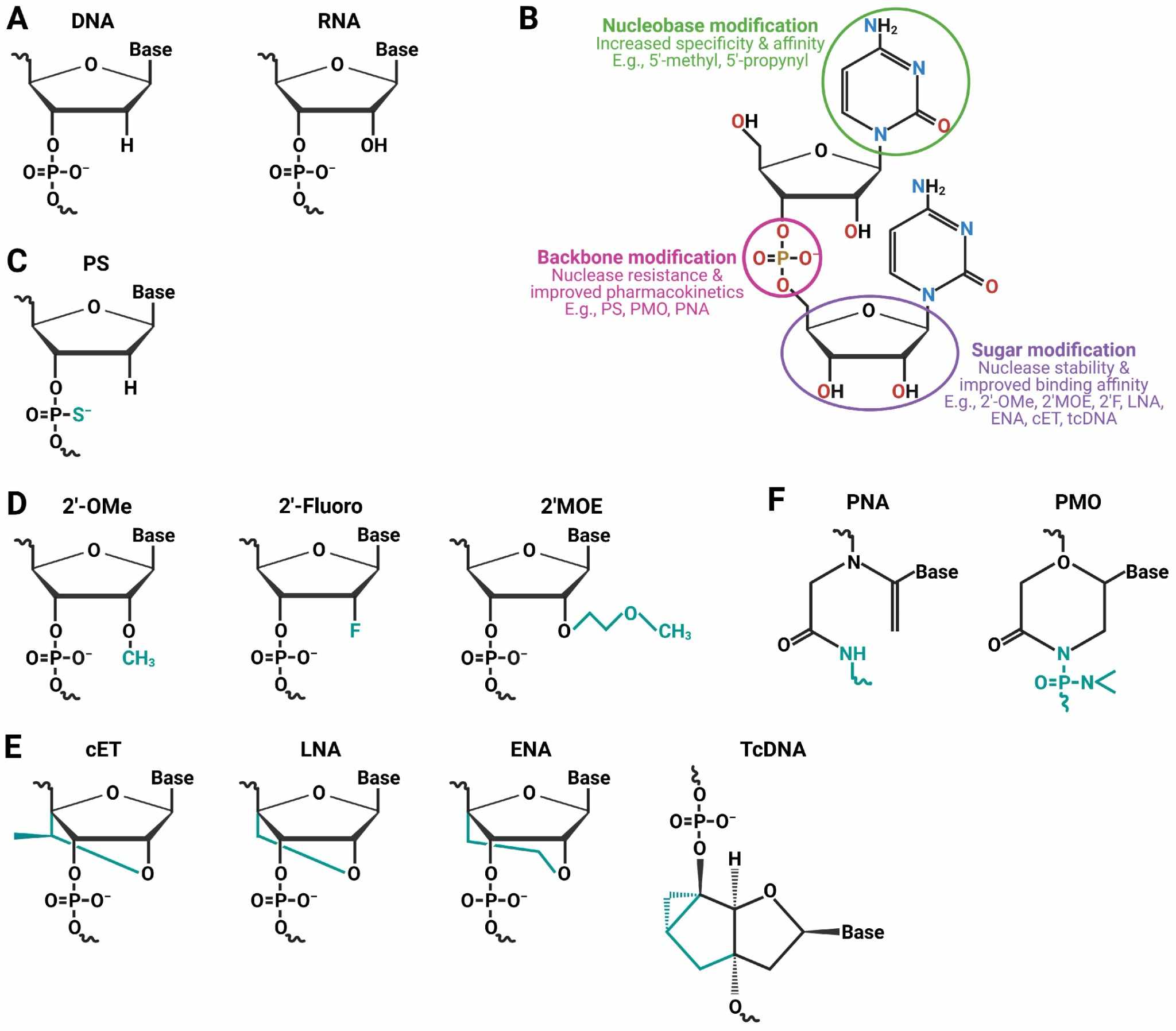 Fig. 2 Some commonly used ON chemistries.2,5
Fig. 2 Some commonly used ON chemistries.2,5
LNA links the 2'-oxygen and the 4'-carbon with a methylene bridge, producing a bicyclic sugar locked in a permanent C3'-endo conformation. The resulting high degree of conformational constraint imparts the largest per-nucleotide increase in melting temperature of any sugar modification. This allows 14–16-mer gapmers to reach the affinity long considered the exclusive domain of 20-mers. The drawback is over-rigidification: fully LNA-modified strands can cause impaired RNase-H cleavage and hepatic accumulation. For this reason, clinical candidates use sparse LNA patches (usually one to three residues) within MOE or 2'-F wings to localise the affinity boost without freezing the entire helix.
cEt methylates the LNA bridge, creating a 2',4'-constrained ethyl bicyclic sugar with the C3'-endo lock retained but a more hydrophobic face. The additional methyl enhances the lipophilicity and membrane permeability and reduces the conformational entropy penalty associated with duplex formation. cEt has been found to have similar melting enhancements to LNA in head-to-head comparisons but is observed to have a "cleaner" safety profile in rodent and primate toxicology studies, which has been postulated to be due to the extra steric bulk hindering non-specific protein binding which can lead to hepatotoxicity or complement activation.
Ethylene-bridged nucleic acids (ENA) substitute the methylene bridge of LNA with an ethylene unit. This additional rotatable bond slightly relaxes the sugar but keeps most of the affinity gain. 2'-O-N-methyl-acetamido (MAA) adds a polar carbonyl that can increase aqueous solubility and cellular uptake through amino-acid transporters. 2'-cyanoethyl incorporates a metabolically soft chain that can be cleaved within endosomes to restore native RNA for transient activity. Each of these edits is currently being evaluated in allele-selective siRNAs or self-amplifying mRNA templates to extend the limits of potency, duration and tissue selectivity.
While LNA and cEt provide the greatest gain in per-nucleotide affinity, the potential for excessive rigidification and hepatic accumulation makes their overuse undesirable. ENA and MAA provide intermediate affinity gains with the benefits of improved solubility and reduced toxicity, but do not yet have decades of clinical experience like MOE. 2'-cyanoethyl provides a cleavable pro-drug option, but also requires validation of esterase activity in the target tissue of each new drug. The new consensus, then, is to use these ultra-constrained sugars judiciously, at one or two residues per strand and embedded within a well-characterized 2'-MOE or 2'-F scaffold in order to create "third-generation" mixmers that can achieve new heights of affinity without losing manufacturability or falling outside regulatory precedent.
Sugar selection is often less a question of choosing the "best" modification, but rather of matching a chemical personality to a therapeutic job description. The 2'-ether that gives you month-long knock-down in liver may over-stabilise a neuronal splice-switcher or trigger complement spikes in kidney. Successful programmes therefore treat the sugar palette as a tunable matrix: lock affinity with LNA where structure is stubborn, soften with MOE where safety is paramount, and sprinkle fluorine only where seed discrimination is non-negotiable.
Potency is determined by the degree to which the sugar fixes the ribose into the C3'-endo pucker: LNA > cEt > 2'-F > 2'-MOE > 2'-OMe. However, ultra-high affinity can over-stabilize duplexes, cause strand dissociation problems within RISC or RNase H, and form stiff backbones that resemble viral RNA and indirectly trigger TLR7/8. Developers thus incorporate high-affinity sugars only at positions where binding energy is limiting (seed region, splice junction or SNP-discrimination hinge) and then use softer 2'-MOE or 2'-OMe on either side to regain breathing room. Safety can also be fine-tuned by modulating phosphorothioate density: each additional sulfur boosts protein binding and complement consumption so the aim is to use the fewest PS possible while still achieving renal retention. Finally, immunogenicity can be reduced by avoiding highly structured high-affinity motifs at the 5'-end, and by capping the termini with 2'-OMe, patterns which have repeatedly lowered cytokine release across multiple clinical programmes.
siRNA can be heavily sugar-substituted as RISC cleavage can occur through an unmodified central wedge; alternating 2'-F/2'-OMe is therefore standard for seeds and wings, while LNA is reserved for one or two "affinity hotspots" to ensure invasion of structured viral genomes. Antisense gapmers face a stricter constraint: the central DNA stretch must remain unmodified to recruit RNase H, so 2'-MOE or cEt wings are used to clamp the target while LNA is positioned only at the gap/wing junction to terminate cleavage precisely. Splice-switching ASOs are fully sugar-modified and must avoid any rigid sugar in the central six-mer to allow spliceosome remodelling; designers therefore embed LNA or cEt at the exon-junction hinge and flank them with 2'-MOE to maintain flexibility. mRNA therapeutics use sugar modification sparingly—only selected uridines and cytosines carry 2'-OMe—to avoid stalling ribosomes, while self-amplifying RNA eschews sugar changes entirely and relies on nucleobase modification for immune evasion. Thus, the same sugar can be either enabling or prohibitive depending on the catalytic context in which it is placed.
Table 2 Modality-Specific Sugar Strategies
| Modality | High-Affinity Sugar | Soft Sugar | Catalytic Constraint |
| siRNA | 2'-F seed, LNA hotspot | 2'-OMe wings | Central wedge must breathe |
| Gapmer ASO | LNA/cEt at gap edge | 2'-MOE wings | DNA gap must stay native |
| Splice-switch | LNA/cEt at junction | 2'-MOE body | Central six-mer must flex |
| mRNA | 2'-OMe sparse | Native | Ribosomal A-site tolerance |
LNA and cEt both rely on bicyclic nucleoside building blocks, which are prepared by low-temperature cyclisation cascades. Yields are moderate and the routes involve anhydrous fluoride or mesylate chemistry, so the capital costs of cryogenic skids and fluoride-resistant metallurgy are greater than for 2'-OMe or 2'-F. In contrast, 2'-MOE ethylene-glycol etherification chemistry uses commodity epoxides and can be performed in normal stainless-steel reactors, so 2'-MOE is the cost benchmark against which all large-scale campaigns are measured. For this reason developers will often do a cost-benefit analysis early in lead optimisation: if a single LNA residue can be used to shorten the ASO by three bases, the savings in raw material cost and downstream purification will often outweigh the higher monomer cost. In contrast, if a lead candidate has to incorporate >30 % LNA density in order to be potent, switching to cEt or mixed 2'-F/2'-MOE patterns will often deliver equivalent affinity at lower cost and with a wider safety margin. Dual-source contracts are negotiated for high-risk sugars (LNA, cEt) while commodity 2'-OMe and 2'-F are single-sourced with agreed change-control windows, so that any upstream route change can be absorbed without repeating pivotal toxicology lots.
Industrial scale manufacture of sugar-modified nucleotides must introduce the alkyl, fluoro or ether substituent into a highly protected ribose, without loss of anomeric purity, protecting-group orthogonality or final amidite crystallinity. Every additional 2'-substituent extends the synthetic tree, increases the solvent load, and contributes new classes of impurities (regio-isomeric sugars, trace metals, residual fluoride) which must be removed before the monomer can be released under cGMP. Success therefore depends on the early choice of protecting ensembles that are robust to both electrophilic fluorination and metal-catalyzed side reactions, and on in-line analytics that monitor fluoride, moisture, and anomeric purity in real time.
Attachment of a 2'-O-methoxyethyl, 2'-fluoro or constrained-ethyl group involves regioselective chemistry that leaves the 3'-position open for later phosphitylation. Classical routes use transient bis-silyl or orthoester masks that only deprotect the 2'-hydroxyl, the subsequent alkylation or fluorination step is then run under rigorously anhydrous conditions since ppm-level moisture hydrolyses activated sugars and generates mixed 2',3'-substituted side products that co-crystallize with the desired isomer and are not detected by routine UV assays. Protecting groups must therefore be orthogonal: acid-labile DMT for 5'-OH, base-labile amides for nucleobase amines, and fluoride-labile silyls for 2'-OH, so that the modification step can be performed before base-protection without cross-reactivity. Failure to maintain this firewall results in premature detritylation or acyl migration that seeds truncated phosphoramidites impossible to purge downstream. Recent flow-chemistry protocols mitigate these risks by mixing the fluorinating reagent and protected sugar in a cooled plug-flow tube, minimizing local HF accumulation and enabling telescoped work-ups that avoid intermediate isolation.
The specification for the qualified monomer should be such that it contains only one 2'-epimer and is free of regio-isomeric sugars, residual fluoride and moisture above the level which hydrolyses the phosphoramidite under long term storage conditions. Such specification limits are justified by forced-degradation studies in which the protected nucleoside is stored at elevated temperature under 75 % relative humidity to show that protecting-group integrity does not exceed acceptance criteria for a period of at least 12 months. Water content is determined by Karl-Fischer coulometry which is carried out under an inert gas atmosphere; a dual limit (closed-vial ppm and in-use ppm) is designed to reflect typical factory-floor exposure during pneumatic transfer. Residual fluoride can be determined by ion chromatography with suppressed conductivity; any ion above the ppb-level has the potential to catalyze silyl migration and/or phosphate oxidation during subsequent coupling cycles. A stability-indicating HPLC method is based on two columns (ion-pair and hydrophilic interaction) which can resolve authentic side-products, e.g. 3'-OMe or 3'-F isomers; new peaks which appear above 0.1 % area under stress conditions will lead to re-validation of the synthetic route. The monomer is finally packaged in aluminium-laminated pouches, back-flushed with argon, and overwrapped with desiccant packs; real-time ingress of moisture is monitored by near-infrared spectroscopy to ensure that critical water activity stays below validated ceiling throughout labelled shelf-life.
Multi-ton campaigns of sugar-modified nucleosides are done in continuous-flow or loop-type reactors, usually anhydrous to prevent any chance of anomericization, with good heat transfer capability to remove the exotherm from metal-hydride deprotonation or electrophilic fluorination. Heat removal in batch-mode reactors above 2 m³ volume is often the rate-limiting step, which tends to form local hot-spots that may be harsh enough to anomerise the sugar or cause silyl ether cleavage, so plug-flow tubular reactors with inline static mixers are preferred for the modification step. The work-up steps—quench, extract, crystallize—are done in a train of stainless-steel reactors qualified for explosive-proof operation as methyl iodide or DAST vapors can build up under the nitrogen blanket used to prevent moisture uptake in the headspace. Recovery of solvents is built in: THF is distilled under nitrogen and passed through molecular-sieve columns for immediate reuse to lower both cost and environmental impact. Change-control governance is in place for the synthetic route, crystallization solvent and drying protocol, each locked under version control with full comparability study required if any parameters are changed (e.g. new filter aid or different dryer temperature). The formal comparability study includes side-by-side phosphitylation to demonstrate that amidite yield and impurity profile are within validated ranges. The in-process checkpoints (fluorination endpoint by 19F-NMR, moisture after vacuum drying, metal content before release) are captured in the GMP batch record, so that each lot can be fully traced back to raw-material receipts and forward to the final oligonucleotide drug substance.
We provide a comprehensive range of 2'-MOE and other sugar-modified nucleotide solutions designed to support the development of next-generation oligonucleotide therapeutics. By combining advanced modification chemistry with high-quality manufacturing and technical expertise, we help customers improve oligonucleotide stability, potency, and pharmacokinetic performance while maintaining efficient and scalable synthesis processes.
Our portfolio includes a full set of 2'-O-methoxyethyl (2'-MOE) nucleotides covering all canonical bases, as well as other advanced sugar modifications used in modern oligonucleotide drugs. These sugar-modified nucleotides are widely applied in antisense oligonucleotides and emerging RNA modalities to enhance nuclease resistance, binding affinity, and in vivo durability. All products are manufactured to high purity standards with strict control over impurities and moisture, ensuring reliable performance during oligonucleotide synthesis. Our broad modification offering allows developers to tailor sugar chemistry to specific therapeutic and delivery requirements.
To enable precise and efficient oligonucleotide assembly, we supply protected sugar-modified nucleoside monomers that are fully compatible with phosphoramidite chemistry and automated solid-phase synthesis. These monomers incorporate optimized, orthogonal protecting group strategies designed to support controlled reactivity and clean deprotection. Our protected 2'-MOE and sugar-modified monomers deliver high coupling efficiency, minimal side reactions, and consistent sequence fidelity, even in complex or heavily modified oligonucleotide constructs. This synthesis-ready design supports robust manufacturing at both development and commercial scales.
For programs requiring specialized sugar modifications or unique design strategies, we offer custom development and process optimization support. Our technical team assists with modification selection, synthetic route development, protecting group optimization, and impurity control to ensure that custom sugar-modified nucleotides are scalable, reproducible, and suitable for long-term supply. By integrating modification design with manufacturability considerations early in development, we help customers reduce technical risk and accelerate progression toward clinical and commercial manufacturing.
We provide GMP-ready supply options for 2'-MOE and other sugar-modified nucleotides to support clinical and commercial oligonucleotide manufacturing. Our materials are accompanied by clear specifications, certificates of analysis, traceability, and change control documentation, aligning with regulatory and quality expectations. This regulatory-focused approach enables smooth transitions from development to GMP production while ensuring consistent quality and long-term supply continuity.
If you are developing next-generation oligonucleotide drugs that require advanced sugar modifications such as 2'-MOE, our team is ready to support your program. Contact us today to request technical information, samples, or quotations, or to discuss custom sugar-modified nucleotide solutions and GMP-ready supply options tailored to your development needs.
References
A 2'-O-methoxyethyl substitution improving stability and potency.
Primarily in antisense oligonucleotides.
It provides stronger binding and longer tissue retention.
Yes, including LNA and cEt modifications.
Yes, but they significantly improve drug performance.


 Loading ......
Loading ......