Metabolically stabilizing RNA into a therapeutic scaffold can be achieved by replacing the 2'-hydroxyl with fluorine. The C–F bond is metabolically inert, while the high electronegativity of fluorine skews the sugar ring into a C3'-endo pucker that condenses duplex geometry and increases melting temperature. The atomic radius of fluorine is similar to hydrogen so 2'-F nucleotides are accommodated in polymerase active sites; densely substituted without loss of base-pair fidelity or translational efficiency. The modification is now codified in every clinically approved siRNA, as well as the wings of antisense drugs, validating 2'-fluoro as the minimal, most effective chemical edit for systemic RNA therapeutics.
Endogenous RNA is degraded by the ubiquitous ribonucleases within minutes. It can also stimulate toll-like receptors and is quickly cleared by glomerular filtration in the kidney. This is not the characteristic of a substance suited to systemic therapy. Chemical modification is, therefore, not an academic embellishment but an engineering imperative that transforms a labile signalling molecule into a drug-like biopolymer with the capacity to access intracellular targets at manageable doses and dosing intervals.
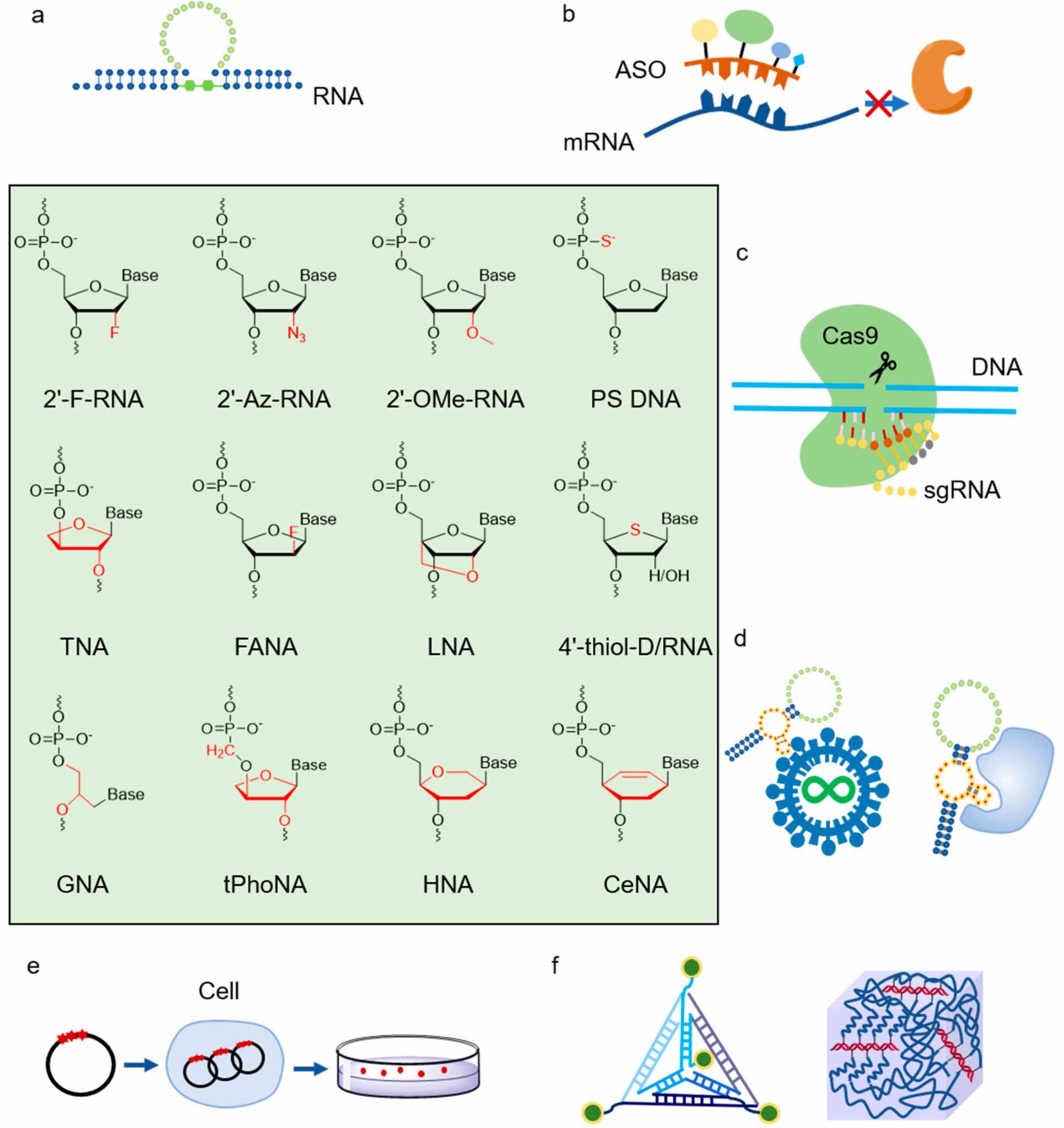 Fig. 1 Application of XNAs and XNAPs.1,5
Fig. 1 Application of XNAs and XNAPs.1,5
The 2'-hydroxyl group serves as an internal nucleophile, attacking the adjacent phosphate and causing strand scission under physiologic magnesium and pH. Simultaneously, TLR7/8 and RIG-I sense the unmodified ribose pattern and initiate a type-I interferon response that clears the transcript and causes inflammatory side effects. Rapid renal filtration also further reduces its half-life, so that even with high-dose infusions, it is difficult to maintain an effective concentration in tissues.
Sugar, backbone and base edits have been introduced to hide elements that can be used to detect a therapeutic. 2'-substitutions delete the reactive hydroxyl, phosphorothioate linkages substitute vulnerable oxygens with sulfur and base analogues, such as pseudouridine, can avoid innate sensors while maintaining fidelity. The edits can be combined in a positional way that optimizes stability, affinity and immunogenicity, forming a modular chemical grammar that extends to siRNA, antisense and mRNA modalities.
Of 2'-substituents, fluorine has the smallest steric profile with the largest electronic effect. The C–F bond is unhydrolyzable and unoxidizable, and its high electronegativity pre-organizes the sugar into a C3'-endo conformation that raises melting temperature without inhibiting RNase-H or ribosome function. 2'-F is in clinical programs at dose densities where the bulkier ethers would be toxic, enabling ultra-stable siRNA and long-half-life antisense drugs that can be dosed monthly.
Table 1 Comparative stability profile of native vs 2'-fluoro RNA
| Feature | Native RNA | 2'-Fluoro RNA |
| 2'-Substituent | –OH | –F |
| Nuclease resistance | Low | Very high |
| Sugar pucker | C2'-endo ↔ C3'-endo | Locked C3'-endo |
| Tm elevation | Baseline | High |
| Immunogenicity | High | Very low |
| Clinical status | Pre-clinical | Approved drugs |
Replacing 2'-hydroxyl with fluorine atom results in minimal steric change and introduces the maximal electronic perturbation possible with a single atom. The C–F bond is short, chemically inert and highly polar, which pulls electron density away from the ring oxygen and locks the sugar in a C3'-endo conformation that mimics the native A-form helix. Because fluorine's van der Waals radius is only marginally larger than hydrogen's, 2'-F nucleotides fit polymerase active sites without steric clash, yet their electronegativity pre-organizes the backbone for tighter base stacking and higher melting temperature. The modification therefore converts RNA from a labile signalling molecule into a drug-like scaffold that resists both enzymatic and chemical degradation without sacrificing decoding fidelity.
The modification is typically made on a transiently protected ribose: 3',5'-bis-silyl or orthoester masks are installed, the 2'-alkoxide is formed with a strong base, and electrophilic fluorine is delivered by DAST, XtalFluor-E or Selectfluor under rigorously anhydrous conditions. The procedure is moisture sensitive: trace water hydrolyses the activated sugar and gives 2',3'-mixed fluorides that co-crystallize with the desired isomer and are not seen by routine HPLC. Because the C–F bond is inert to acid, base and oxidation, the modification is stable through all subsequent protecting-group manipulations—acid detritylation, base amide cleavage and phosphate oxidation—without fragmentation or epimerization and is the most robust single-atom edit known to oligonucleotide chemists.
High-resolution crystal structures have shown that 2'-F strongly biases the furanose ring conformation towards a near-ideal C3'-endo conformation. This results in shortening of the phosphate-to-phosphate rise and a deepening of the major groove. This pre-organization of the sugar conformation imposes a lower entropic penalty upon duplex formation, resulting in increased melting temperatures and tighter binding to complementary RNA or DNA. The fluorine atom can also polarise the nucleobase, leading to increased strength in Watson-Crick hydrogen bonds, as well as improved π-stacking without base-pair geometry distortion. The same pucker bias in a single-stranded context can discourage non-canonical folds such as G-quadruplexes, ensuring that the strand remains extended and accessible for RISC loading or RNase-H recognition.
Relative to native RNA, 2'-F imparts the greatest increase in nuclease resistance with the smallest steric bulk. Relative to 2'-O-methyl, 2'-F has a higher melting temperature and better chemical inertness, but lacks a hydrogen-bond acceptor to enhance protein recognition at times. Versus 2'-MOE, 2'-F is easier to install and crystallizes more readily, but is not as sterically shielded from exonucleases, so many clinical gapmers are comprised of 2'-F at hotspot positions combined with 2'-MOE in terminal wings. Against locked nucleic acid (LNA), 2'-F provides similar affinity increases without the conformational rigidity that can negatively impact RNase-H cleavage, and is the preferred modification when catalytic turnover is desired.
2'-fluoro nucleotides stabilize RNA by three mutually reinforcing mechanisms: they eliminate the intramolecular transesterification route for cleavage of native RNA, they impose a steric/electronic barrier to exo- and endonucleases and they pre-organize the sugar in a high melting C3'-endo conformation. The contributions of these three effects are additive across the strand, resulting in multi-log serum half-life extension without increasing the sequence length or synthetic complexity.
In addition to loss of hydroxyl, the fluorine atom is itself directed into the minor groove, where it exerts a local dipole field that sterically repels the positively charged active-site residues of nucleases. For instance, the crystal structures of RNase A demonstrate that a protonated 2'-OH is needed for the first step in the transphosphorylation mechanism; the lack of a suitable proton donor in 2'-F RNA therefore acts as a dead end for the catalytic process. 3'-exonucleases also depend on a magnesium coordinated water molecule attacking the scissile phosphate; the inductive effect of fluorine reduces the basicity of the 3'-oxygen, which in turn reduces the binding affinity of the catalytic metal ion and results in cleavage being slowed down by several orders of magnitude. The protection is also absolute against serum endonucleases and cellular RNases, meaning that the oligonucleotide can transverse extracellular spaces, endosomal vesicles and nuclear speckles without detectable degradation. The steric effect of fluorine is only slightly larger than that of hydrogen, so high density substitution does not inhibit hybridisation or strand invasion of structured RNA elements such as stem-loops or G-quadruplexes, and this property is exploited in allele-selective gapmer designs.
Native RNA is also sensitive to backbone cleavage under mildly alkaline conditions by a process known as β-elimination, which begins with deprotonation of the 2'-OH; the resulting alkoxide then attacks the adjacent phosphate group to produce a 2',3'-cyclic phosphodiester that cleaves the backbone. Fluorine is not sufficiently acidic and so the reaction manifold is eliminated entirely. In a similar manner, acid-catalyzed depurination is lessened as the electron-withdrawing fluorine reduces the electron density at N9 and the glycosidic bond is therefore less prone to protonation. Oxidative stress, such as that created in inflammatory micro-environments, can also generate reactive oxygen species that are able to abstract hydrogen from the 2'-carbon; the C–F bond, on the other hand, is resistant to such radicals and abasic sites that would initiate backbone cleavage cannot form. These chemical protections increase the shelf life of the oligonucleotide from hours to years and permit slow-release implants as well as lyophilized formulations that can be stored at room temperature without degradation of potency.
The conformational lock is propagated over the full length of the helix: adjacent sugars are forced into the same C3'-endo envelope, resulting in a cooperative stacking network that raises the enthalpy of duplex formation. The entropic penalty upon hybridization is thus ameliorated, and 19-mer siRNAs are able to match the melting temperature of 21-mer native duplexes. This reduction in length also reduces manufacturing costs and minimizes off-target seed matches. In addition to shielding the ribose, the fluorine atom polarizes the nucleobase, enhancing hydrogen-bond donor and acceptor interactions without perturbing the canonical Watson-Crick geometry. in vivo, the increased Tm results in slower dissociation from target mRNA, lengthening the residence time of the ASO-RNA heteroduplex and thus the time window for RNase-H cleavage or splice-switching activity. Finally, the enhanced thermal stability allows for the use of higher annealing temperatures during formulation, lowering the risk of aggregate formation and improving the monodispersity of lipid-nanoparticle or GalNAc-conjugated formulations.
The minimal modification that can be introduced to an siRNA while still maintaining activity is 2'-fluoro (2'-F) nucleotides. 2'-F is the most activity-retentive chemical optimization of siRNA, rendering them nearly nuclease proof, while still preserving the stereochemistry necessary for loading and cleavage by RISC. The small size of fluorine, being only slightly larger than hydrogen, allows for extensive substitution without incurring a steric burden. This results in the formation of ultra-stable duplexes which are able to withstand systemic circulation, but are still able to unwind easily inside of Argonaute. The 2'-F edit is now incorporated into all clinically approved siRNAs, establishing 2'-F as the benchmark sugar edit for siRNA therapeutics.
The distribution of 2'-F is not uniform. A fully fluorinated passenger strand is sterically and chemically inert to RISC. This precludes loading of the passenger into Argonaute, leaving the guide strand as the only contender for loading, and avoiding the competition that would otherwise lead to a loss in silencing potency. The distribution of fluorine on the guide strand is not random but also functionally partitioned. The seed region (position 2–8) is rich in fluorine that its electronegativity constricts base-pair geometry and heightens single-base mismatch discrimination. The cleavage center (position 10–11), by contrast, remains mostly native or modestly methylated to avoid conformational breathing that is essential for endonucleolytic cleavage. The region-specific mosaic is borne out by deep-sequencing experiments that demonstrate that passenger-strand-derived off-target effects are reduced by more than an order of magnitude by asymmetric 2'-F patterns without sacrificing the on-target knock-down window of its unmodified parent sequence. The 3' overhangs of both strands are fully fluorinated. This steric shield against 3'-exonucleases prolongs circulation half-life from minutes to days and allows for once-monthly sub-cutaneous dosing regimens that have become industry standard for GalNAc-conjugated siRNA therapeutics.
The energetic advantage provided by the C3'-endo lock has been demonstrated to result in increased RISC loading rates and a more complete mRNA cleavage. While 2'-F-rich siRNAs can reach maximal liver target knock-down levels in vivo after 24 hours, lightly modified duplexes require several doses over the course of several days to achieve comparable levels of mRNA reduction. The modification also increases the stability of the RISC particle itself, increasing the half-life of the guide strand within RISC from hours to days, and resulting in an overall extended period of gene silencing which can last for weeks after a single sub-cutaneous injection. Finally, the increased thermal stability of the duplex allows for shorter guide strands (19–21 mer) to be used that are still able to bind with high affinity, lowering manufacturing costs and minimizing the risk of non-specific hybridisation to partially complementary transcripts.
Off-target silencing is largely entropy driven by partial complementarity in the seed region of the guide strand. Placing 2'-F at key seed positions increases the enthalpic penalty of bulged or mismatched hybrids, tipping RISC towards fully complementary targets. Genome-wide microarray analysis show that a single 2'-F substitution in position 7 of the guide strand abolishes >50% of seed-dependent off-target signatures without impacting on-target cleavage efficiency. In addition, the fluorine atom occludes the minor-groove face that is recognized by TLR7/8, eliminating the cytokine burst (IFN-α, TNF-α) that can result from high-dose unmodified siRNA. The dual benefit of thermodynamic discrimination and immune quiescence has made 2'-F the default edit for clinical candidates, permitting developers to comfortably surpass the strict off-target thresholds established by regulators while maintaining the strong gene-silencing potency needed for therapeutic efficacy. Finally, the small steric size of fluorine ensures that dense substitution does not negatively impact hybridization or strand invasion of structured RNA elements such as stem-loops or G-quadruplexes, a property that is exploited in allele-selective siRNA designs where single-nucleotide discrimination is needed for therapeutic selectivity.
2'-fluoro nucleotides convert ASOs into high affinity, nuclease-resistant therapeutics that utilize catalytic and steric-block mechanisms of action. The small steric bulk of the modification does not compromise RNase-H recruitment. Electronic effects associated with this modification result in a tighter duplex geometry and allow for shorter, more sequence specific oligonucleotides to reach intracellular RNA targets at an achievable dose and dosing frequency.
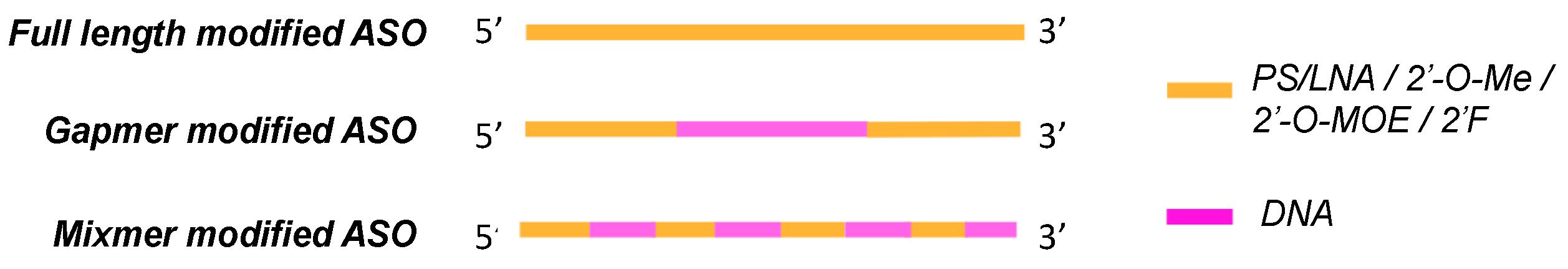 Fig. 2 Antisense oligonucleotide (ASO) design.2,5
Fig. 2 Antisense oligonucleotide (ASO) design.2,5
Molecular-dynamics simulations show that the fluorine dipole is aligned along the helical axis, producing a local electric field that stabilizes the anti glycosidic conformer and disfavors syn rotamers, which would expose the bases to solvent. This pre-organization decreases the entropic cost upon duplex formation and thus permits sub-nanomolar occupancy of viral or oncogenic RNAs that adopt stable secondary structures. In gapmer designs, the 2'-F is usually enriched in the 5' and 3' wings while a central DNA gap is retained as a flexible "trigger" for RNase-H cleavage; fluorinated clamps then prevent "breathing" that would otherwise permit the target RNA to escape and re-fold. The gain in affinity is greatest in RNA: DNA heteroduplexes where the C3'-endo sugar mimics the native A-form geometry and creates a seamless fit along the major groove. Crucially, the higher binding energy does not come at the cost of strand invasion of structured RNA elements like stem-loops or G-quadruplexes, so that ASOs are now able to target previously "undruggable" parts of the transcriptome. Finally, the higher melting temperature buffers against single-base mismatches and thus tightens the therapeutic index and lowers the dose needed to achieve maximal mRNA reduction.
Table 2 Affinity Gain Across ASO Architectures
| Architecture | 2'-F Pattern | Tm Gain vs DNA | Functional Outcome |
| Gapmer wings | Alternating 2'-F/2'-OMe | Moderate | Shorter gap, less off-target |
| Steric-block | Full 2'-F | High | Invades stem loops |
| Mixmer | 2'-F seed, 2'-OMe flanks | High | Allele-selective bias |
The versatility of 2'-F arises from its ability to deliver high affinity without the conformational rigidity that can impair catalytic turnover. In gapmer applications, the central gap of 8–10 DNA nucleotides provides the flexibility required for RNase-H to engage the heteroduplex, while the 2'-F wings maintain a high local melting temperature that prevents the target RNA from dissociating before cleavage occurs. For steric-block or splice-switching modalities, fully 2'-F backbones are preferred because they do not recruit RNase-H and therefore avoid unintended target destruction. The modification's small size ensures that the ASO can invade structured pre-mRNA regions such as splice enhancer or silencer elements, displacing spliceosomal proteins and redirecting exon inclusion or exclusion. Recent clinical candidates exploit this mechanism to redirect splicing in Duchenne muscular dystrophy, spinal muscular atrophy and inherited blindness, where single-base precision is essential for restoring the reading frame without creating new cryptic splice sites. Finally, the compatibility of 2'-F with both catalytic and occupancy-based mechanisms allows developers to toggle between degradation and splice correction using the same chemical platform, simplifying regulatory pathways and manufacturing logistics.
The methyl-like size of fluorine introduces a modest but meaningful increase in logP, facilitating reversible binding to albumin and α-2-macroglobulin that buffers the oligonucleotide against immediate glomerular filtration. This protein association creates a depot effect that sustains plasma concentrations above the efficacy threshold for days after a single sub-cutaneous injection. Once in tissue, the 2'-F sugar resists both exo- and endonucleases, enabling the ASO to traverse the interstitial matrix and reach target cells in liver, muscle and CNS compartments. The modification does not trap the molecule in endosomal compartments; instead, it facilitates release into the cytosol and nucleus, where it can engage mature mRNA or nascent pre-mRNA transcripts. The broad biodistribution underpins the clinical success of 2'-F-rich gapmers in hepatic, neuromuscular and ocular indications, where durable gene modulation is achieved with quarterly or even semi-annual dosing intervals. Finally, the enhanced stability reduces the need for high phosphorothioate content, lowering the risk of protein-binding-related toxicities such as complement activation or platelet aggregation, and enabling higher cumulative doses for chronic genetic diseases.
To develop a clinically actionable siRNA or antisense molecule, the same chemical modification must simultaneously improve serum half-life, affinity to target, and also avoid detection by the innate immune system. 2'-fluoro provides this 3-way hit by fixing the sugar moiety in a high melting C3'-endo configuration while maintaining a minimal steric profile. The challenge is spatial precision: fluorine must be sufficiently ubiquitous to disarm nucleases, but not so dense as to disrupt catalytic scope, trigger immune recognition and/or drive up manufacturing cost. The sections below cover how developers tune fluorine frequency, mix with orthogonal chemistries, and ensure the right balance is achieved at the in vitro, in vivo, and cGMP scales.
While 2'-F is less sterically demanding than 2'-OMe, its electronegativity is still detectable by TLR7/8 when introduced at every position. Clinically, fully 2'-F modified duplexes lead to a transient IFN-α response in primates, but a mixed pattern of 2'-F in the seed and 2'-OMe elsewhere in the duplex can eliminate the cytokine signature without loss of potency. These effects are even more marked when fluorine modification is restricted to the guide strand seed and the passenger strand is completely methylated, a combination that conceals the minor-groove pattern targeted by pattern-recognition receptors without affecting duplex stability.
Density is adjusted by design mechanism. RNase-H-dependent gapmers have evolved to tolerate 30–40 % 2'-F in the wings in order to maintain a flexible DNA gap while still having an acceptable melting temperature, while splice-switching mixmers rarely go over 50 % due to more serious concerns about over-rigidifying the helix. Density is also inversely related to dose: a 60 % fluorinated 16-mer can reach the same level of liver knock-down as a 20-mer with only 30 %, but the shorter sequence has lower cost and will leave behind fewer off-target seeds in the genome. Algorithms therefore use free-energy penalties for each 2'-F in order to predict the minimal fluorine load that can still achieve the Tm and serum-stability requirements, a strategy that has so far resulted in multiple approved drugs dosed once monthly.
2'-F is infrequently used as a monotherapy. Mixing with 2'-OMe in the seeds of siRNA duplexes can provide a mixture of immune silence and high affinity, and alternating with phosphorothioate linkages in the termini of siRNAs or gapmers can provide increased serum protein binding without reintroducing pro-inflammatory sulfur density. Adjacent base analogues such as 5-methyl-cytosine or 2-thio-uracil can further increase Tm and diminish off-target seeds, enabling the development of ultra-short (15-mer) gapmers without loss of potency. The design rationale has been corroborated by orthogonal analytics, including 19F-NMR to quantify fluorine content and LC-MS to show that blended backbone modification patterns do not generate additional diastereomers or mutagenic impurities during long-term storage.
Synthesizing 2'-fluoro nucleotides at scale thus involves trading off the smallest possible atom with the largest possible swing in chemical reactivity. Fluorination needs to take place on a heavily protected ribose under rigorously anhydrous conditions, but the final monomer needs to survive oxidative iodine baths, acid detritylation and global ammonia cleavage without degradation of stereochemistry or build-up of trace fluoride that might poison downstream amidation. Success therefore requires early selection of protecting ensembles compatible with both electrophilic fluorination and metal-catalyzed side reactions, in addition to in-line analytics to monitor fluoride, moisture and anomeric purity in real time.
The key transformation is the regioselective fluorination at C2' following transient masking of 3'- and 5'-hydroxyls. Classic batch routes use DAST or XtalFluor-E under anhydrous conditions; both reagents liberate HF in situ, so even at ppm levels moisture hydrolyses the activated sugar and generates 2',3'-difluoro byproducts that co-crystallize with the desired isomer and are invisible to routine UV assays. Yield erosion is non-linear: a 5 % loss per step that is tolerable at 50 g becomes a multi-kilogram shortfall once 100 kg of starting sugar is committed, forcing campaign extensions that exhaust plant capacity and delay downstream oligonucleotide production. Recent flow-chemistry protocols mitigate this risk by mixing the fluorinating reagent and protected sugar in a cooled plug-flow tube, minimizing local HF accumulation and enabling telescoped work-ups that avoid intermediate isolation. Another complexity arises from the need to maintain the β-anomer: fluorination of a glycal intermediate can produce α/β mixtures that must be separated by crystallization or chromatography, steps that become rate-limiting at ton scale. Finally, the reaction is sensitive to metal impurities: Fe3+ or Al3+ catalyze silyl ether cleavage and generate fluoride complexes that co-precipitate with the product, so stainless-steel reactors must be passivated or lined with PTFE to avoid trace-metal excursions.
Fluorination precedes base-protection, because the strong base needed to generate the 2'-alkoxide causes acyl migration of benzoyl groups if they are already in place. A standard sequence places fluoride on a 3',5'-bis-TBDMS ribose and then strips the silyl ethers in fluoride ion conditions that do not disturb the new C–F bond. Base amides (benzoyl, isobutyryl) are added next under mild Schotten–Baumann conditions that do not epimerize the anomeric centre. Orthogonality is vital: acid-labile DMT must survive the HF generated during fluorination, while fluoride-labile silyls must be stable during the subsequent base-protection step. Any breach of this firewall causes premature detritylation or loss of silyl protecting groups that seeds truncated phosphoramidites impossible to remove downstream. Protecting groups are validated by forced-degradation studies where each mask is exposed to the worst conditions of the next step. The appearance of any new peak above 0.1 % area triggers a route redesign before the chemistry is locked under change-control.
The monomer should contain a single 2'-epimer and must not contain any regio-isomeric fluorides. All other by-products of the DAST reaction, and moisture content above the level required to hydrolyze the phosphoramidite during the stated shelf-life should be at specified levels or less. These limits can be shown by accelerated ageing studies, in which the protected nucleoside is stored at an elevated temperature in an atmosphere of 75 % relative humidity and it is shown that the fluoride and the protecting groups are still within the specified limits after at least one year. Moisture content is determined by Karl-Fischer coulometry under inert gas: the two-tier limit (closed-vial ppm and in-use ppm) simulates exposure on the factory floor during pneumatic transfer. Residual fluoride content is determined by ion chromatography with suppressed conductivity: even ions present at ppb levels have the potential to catalyze silyl migration or phosphate oxidation in the following coupling cycles. A stability-indicating HPLC method using two columns (ion-pair and hydrophilic interaction) resolves any authentic side-products like 3'-F isomers or base-acyl adducts; new peaks above 0.1 % area under stress conditions should lead to re-validation of the synthetic procedure. The monomer is then packaged in aluminium laminated pouches back-flushed with argon and over-wrapped with desiccant packs, and real-time moisture ingress is monitored by near-infrared spectroscopy to ensure that the critical water activity remains below the validated ceiling throughout the stated shelf-life.
We provide a specialized portfolio of 2'-fluoro (2'-F)-modified nucleotides supported by deep technical expertise in oligonucleotide chemistry. Our solutions are designed to help developers enhance RNA stability while maintaining biological activity and manufacturability across siRNA and antisense oligonucleotide programs.
Our portfolio includes high-purity 2'-fluoro-modified nucleotides covering all canonical RNA bases. These materials are widely used in therapeutic oligonucleotides to improve nuclease resistance, duplex stability, and overall RNA durability without significantly increasing steric bulk. All 2'-F nucleotides are manufactured under stringent quality controls to ensure consistent purity, low moisture content, and well-defined impurity profiles. This level of control is essential for reproducible synthesis performance, particularly in sequences where stability is a critical design parameter.
To support efficient chemical synthesis, we supply protected 2'-fluoro nucleoside monomers that are fully compatible with phosphoramidite chemistry and automated solid-phase oligonucleotide synthesis. These monomers incorporate optimized, orthogonal protecting group strategies that enable precise stepwise chain assembly. Our protected 2'-F monomers are engineered to deliver high coupling efficiency, predictable reactivity, and clean deprotection, helping manufacturers minimize side reactions and maintain high sequence fidelity in highly modified RNA constructs.
For programs with unique modification patterns or advanced design requirements, we offer custom synthesis and scale-up support for 2'-fluoro nucleotides. Our technical team assists with synthetic route development, protecting group optimization, impurity mitigation, and process robustness. By addressing manufacturability early in development, we help ensure that custom 2'-F nucleotides are scalable, reproducible, and suitable for long-term supply, reducing technical risk as programs move toward clinical and commercial manufacturing.
We provide GMP-ready supply options for 2'-fluoro nucleotides to support clinical and commercial production of RNA therapeutics. Materials are supplied with clear specifications, certificates of analysis, traceability, and change control documentation, enabling alignment with regulatory and quality expectations. This regulatory-focused supply strategy supports smooth transitions from development to GMP manufacturing while ensuring consistent quality and supply continuity.
If you are developing RNA therapeutics that require enhanced stability through 2'-fluoro modification, our team is ready to support your program. Contact us today to request technical information, samples, or quotations, or to discuss custom 2'-F nucleotide solutions and GMP-ready supply options tailored to your development needs.
References
It increases resistance to nucleases and hydrolysis.
Yes, they are commonly used in guide or passenger strands.
Yes, it stabilizes the RNA duplex structure.
Generally safe when properly designed and dosed.
Yes, often combined with 2'-OMe or base modifications.


 Loading ......
Loading ......