A common artifact of PCR is the formation of primer dimers and nonspecific amplification products. Primer dimers occur when two primers anneal to each other. Nonspecific amplification can occur when primers anneal to sequences other than their target sequences. Both primer dimers and nonspecific amplification products can compete with the desired PCR product for reagents. Additionally, they can cause false positives when assessing PCR product presence. In quantitative PCR, in which absolute numbers of copies of the target are determined based on accumulation of the PCR product during the reaction, primer dimers and nonspecific amplification cause overestimation of the number of copies present. Primer dimer prevention strategies include removal of secondary structure through judicious primer design, optimization of reaction conditions to reduce nonspecific annealing, and hot-start polymerases that are inactive until mixed.
Primer dimers are small artificial DNA fragments formed when forward and reverse primers anneal to each other instead of the target DNA. Exponential amplification of these primer/primer anneals will result in primer dimers. Primer dimers can usually be seen as bright bands of low molecular weight when visualized on a gel. Primer dimers can be distinguished from excess primers because they will be of a higher molecular weight. Non-specific amplification can include primer dimers as well as non-target fragments of DNA that can be amplified by sharing some sequence homology with the primers or can bind to the primers in a process called mispriming. Additionally, structures known as complex multis can also contribute to non-specific amplification. These consist of multimers of primer/primer duplexes. Primer dimers and non-specific amplification products consume primer and enzyme resources that should be utilized on the target sequence. This competition can make it difficult to interpret results and affect the accuracy of quantification.
 Representative gel electrophoresis images of dimer-forming and dimer-free primer pairs from 14 base pair fusion set1,5
Representative gel electrophoresis images of dimer-forming and dimer-free primer pairs from 14 base pair fusion set1,5
Primer dimers occur when two primers anneal to each other and extend from either primer creating a duplex which can be a stable product of amplification that could potentially have a similar structure to a true template and be amplified. Primer-dimers can cause problems because they waste reagents and because they often out-compete the desired target of amplification due to shorter length and kinetics. Additionally, if detection is being done through an intercalating dye or probes, primer dimers will add to the fluorescent signal as would any true product and affect Ct values and apparent quantity of the target. Non-specific products will similarly affect the apparent quantity of the target and can produce bands/artifacts on a gel or melting peaks that overlap with the desired product making it challenging to cleanly separate. Troubleshooting these issues can be difficult and may involve adjusting reaction conditions or designing a new set of primers.
False priming products result from separate thermodynamic and kinetic processes that take advantage of oligonucleotide flexibility. Primer-dimer formation utilizes partial homology at the ends of primers. Often these are G rich ends that can either form G quartets or Watson-Crick type hydrogen bonds that remain stable at lower extension temperatures. Non-specific binding results when primers bind to unintentional sites with adequate homology for the polymerase to begin extension. This is more likely to occur as genomic complexity increases or as target copy numbers decrease. External conditions such as low magnesium concentrations, high concentrations of primers, and increased cycle numbers can also contribute to false priming by decreasing thermodynamic stability needed for true target binding and allowing low stability duplexes to extend.
Table 1 Comparative Characteristics of Primer Dimers and Non-Specific Amplification Products
| Feature | Primer Dimers | Non-Specific Amplification |
| Molecular Origin | Inter-primer hybridization at complementary termini | Promiscuous binding to unintended genomic loci |
| Product Size | Typically 20-60 base pairs | Variable; may approximate or exceed target length |
| Electrophoretic Appearance | Sharp, low-molecular-weight band | Discrete bands or smearing across size ranges |
| Fluorescence Impact | Elevated background in early cycles; reduced assay sensitivity | Variable signal interference potentially mimicking specific products |
| Primary Prevention Strategy | Elimination of terminal self-complementarity; hot-start methodologies | Database screening for specificity; optimized annealing stringency |
Primer dimer occurs when two primers bind to each other instead of binding to the template. They then serve as templates themselves and produce amplicons. Primer dimers occur when the 3' ends of a forward primer and reverse primer are complementary, allowing them to bind to each other. Primer dimers can also occur when there is only one primer and it folds back on itself creating a loop with the 3' ends exposed. Primer-dimer formation occurs because small pieces of DNA can bind loosely to each other if they are complementary. When primer-dimers form, they can easily be converted to permanent binding through polymerization. Primer-dimers will be amplified during PCR often outcompeting the intended target because they are smaller and get amplified quicker.
Hybridization between different primers is the most common cause of dimers. If the forward primer and reverse primer have regions of complementarity at their ends (usually at the 3' ends), the primers can anneal to one another and become double stranded with hydroxyl ends that are available for extension by polymerase. This will form heterodimers, which will then amplify efficiently in PCR since every instance of the forward primer can serve as a template for the reverse primer (and vice versa), regardless of whether there is a DNA target to amplify. Primer dimers become more likely at high concentrations of primers and less likely at high stringency temperatures (higher temps prevent annealing of partially complementary sequences). To prevent primer dimers, redesign the primers so there is no complementary sequence at their ends. You can accomplish this by reshuffling the order of nucleotides or changing nucleotides.
Secondary structures and primer complementarity lead to amplification artifacts. Secondary structure allows primers to form loops in which primer portions anneal to each other. A stem-loop structure will form with sticky ends available for extension. When primer pairs are complementary to each other, hybridization can result in a product that links the forward and reverse primer. Secondary structures are possible due to stable hydrogen bonding between GC content or complementary AT-rich sequences. Unwanted structures will have a ΔG value allowing them to form spontaneously and be stable. By predicting possible secondary structures in primer sequences one can discard potential primers that will self aggregate rather than remaining single stranded.
Non-specific amplification can result from DNA-DNA interactions that allow the polymerase to extend from positions other than the primer annealing sites or from primer-dimer artifacts. These unwanted amplification products will compete with the specific product being amplified for primer, polymerase, and dNTPs. Products that are generated can result from primers that bind to cryptic sites within complex DNA templates, or through reaction conditions that allow extension from mismatches between primers and the template DNA. Such non-specific amplification products reduce the yield of specific product and compete with the specific product during detection, especially if quantitative assessment is being performed. Improper product formation can waste reagents and may also interfere with accurately determining the number of copies present. By knowing what causes these non-specific products to form, strategies can be devised to reduce or eliminate them.
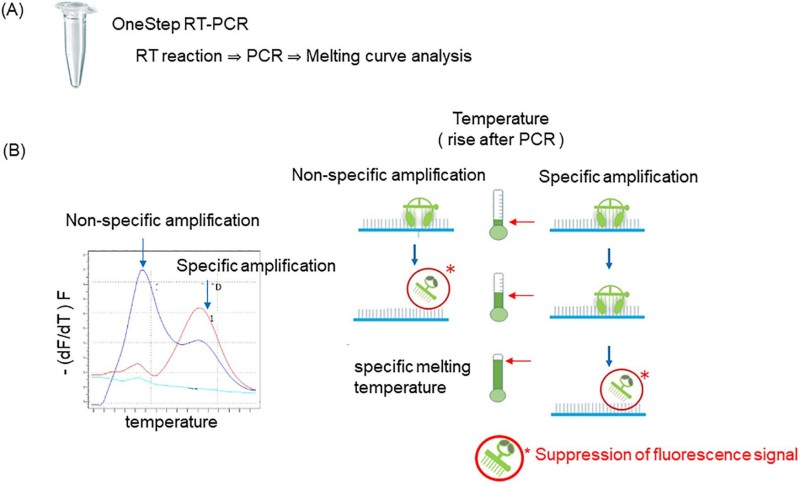 Eprobe mediated RT-qPCR for the detection of leukemia-associated fusion genes2,5
Eprobe mediated RT-qPCR for the detection of leukemia-associated fusion genes2,5
Primers with poor specificity do not have sequences unique enough to selectively amplify only the desired sequence. Cross annealing with similar sequences such as related genes, repeated sequences or conserved sequences that occur in other organisms can occur. Sequence mismatches may not impose enough of a thermodynamic penalty to prevent binding under typical reaction conditions. This problem can be exacerbated if the primer contains long runs of guanine and cytosine. Mis-amplification results in amplification of the unintended product. This can cause the false appearance of the desired product if the unintended product runs at the same size. In applications such as real time quantification, it can create background that makes interpretation of the results impossible.
Mis-optimized thermal cycling conditions leading to relaxed primer/template specificity are a common source of non-specific amplification. Looser conditions allow non-specific binding events with only partial duplex formation to progress to productive amplification. Annealing temperatures may be too low given predicted melting temperatures allowing hybrids with numerous mismatches to stably anneal, while longer times allow for non-specific binding events to attract polymerase. Excessive magnesium concentrations can contribute to relaxed conditions allowing non-specific events to occur more readily by stabilizing mismatches and aiding the extension by polymerase on partially homologous templates. Amplification of these non-specific events can be observed as background haze, additional bands or high baseline in your desired PCR product.
DNA and RNA samples isolated from clinical or environmental samples often contain mixtures of nucleic acids. Low concentration targets may be difficult to detect if they are outnumbered by excess non-target nucleic acids. Non-target DNA and RNA may also contain sequences that the primers can bind to. Degradation of nucleic acids can lead to creation of new primer binding sites. Another issue that can cause primer binding to nonspecific targets is contamination by other substances that affect the kinetics of the PCR reaction. Contamination by DNA from another source, including previous PCR products, people, or other materials can cause primers to bind to these unintended targets. Preventative measures include thoroughly cleaning samples, physically separating pre- and post-PCR reactions, and using enzymes to treat samples that destroy unwanted PCR products but not the oligonucleotides.
Table 2 Etiological Factors Contributing to Non-Specific Amplification
| Causative Factor | Molecular Mechanism | Manifestation in Assay Performance |
| Low Primer Specificity | Promiscuous binding to homologous off-target sequences | Multiple discrete bands; melting curve variants; quantitative inaccuracy |
| Suboptimal Annealing | Insufficient thermal stringency permitting mismatched duplex stability | Background smearing; primer-dimer accumulation; reduced signal-to-noise ratio |
| Template Complexity | Alternative priming sites within degraded or contaminant DNA | Variable background; false-positive signals; irreproducible threshold cycles |
| Component Imbalance | Excessive magnesium or primer concentrations | Enhanced polymerase activity on imperfect substrates; elevated artifact production |
Poor design is one of the reasons for nonspecific amplification. Primer sequences that are too short or have biased base content can increase nonspecific amplification. Thermodynamic properties can also impact amplification specificity when there is a mismatch between the properties of each primer in a pair. If primers are designed without consideration for sequence context, they may bind to hidden sites within the sample or form secondary structures. Secondary structures can hide the 3' end of the primer preventing it from binding to the template. These conditions lead to primers that can bind to each other or other structures rather than to the intended target sequence. This reduces the concentration of primers available for specific target amplification and competes with the desired reaction.
Factors affecting primer length and nucleotide composition can impact PCR specificity. If a primer is too long it can take longer to anneal to its target, and it can bind to sequences with greater mismatches. If it is too short, it might not be unique enough to find its target within the genome. If a primer has too high of a GC content, it will bind too strongly to sequences that are not its intended target. Low GC content can cause a primer to bind weakly to its target. Another factor that reduces PCR specificity is if there is a large difference in melting temperatures between the forward and reverse primer. If one primer has a much higher melting temperature than the other, it will bind first, and might bind to itself, or another piece of DNA, before the other primer is available to bind.
Primers and probes that bind to regions of the genome with repeats or are targeted to genes that have similar family members can create issues with specificity that make amplification more likely to create artifacts. Repeats such as microsatellites or low complexity regions are present throughout the genome and create more sites where primers can bind nonspecifically. If the target gene shares a conserved domain with other genes or if the probe targets a conserved region, then primers may anneal to multiple locations creating mixed amplicons that cannot easily be told apart from the true amplicon. Regions with repeats can slip through bioinformatic checks that look for sequences that perfectly match other regions of the genome. If the primer has partial homology to another region and the reaction conditions are not stringent, the DNA polymerase can extend from the primers.
Synthetic oligonucleotides produced chemically (most common method of synthesis is solid-phase phosphoramidite chemistry) are mixtures of products. During synthesis one nucleotide is added at a time and this reaction is inefficient, leading to incomplete molecules missing a nucleotide at the end. Additionally there are side products from the synthesis such as leftover protecting groups, activators etc. that are not removed during the cleavage from the solid support and deprotection steps. Unless these incomplete sequences and impurities are chromatographically separated from the desired sequence, false positive signals will occur. This is because the incomplete sequences will still be able to bind to the polymerase (though with lower affinity) and emit fluorescence during the detection step. This leads to a lower signal to noise ratio, which decreases the sensitivity of the assay and causes variations in quantification that are unreproducible.
Fragmented products are generated as a natural consequence of stepwise chain growth and will be present alongside complete strands in a synthetic reaction mixture. This phenomenon occurs when one or more activated phosphoramidites do not successfully couple with a growing chain (resulting in a pool of molecules with the same end group configuration but missing interior residues). Their presence is undesirable as they will bind to template strands, but will not be able to be efficiently elongated by polymerase. This effectively removes active enzyme from the reaction as it becomes bound to truncated products. Another source of fragments stems from deletion products caused by unsuccessful oxidative formations within the DNA backbone. These fragments have different sequences than the desired full-length product and could bind elsewhere on the genome.
By chromatographic purification, full-length product can be separated from truncated sequences that result during synthesis. Chromatography also yields a product population that is compositionally pure enough to perform consistent hybridization to the target sequence. Purification separates incomplete reaction products and residual reagents that could contribute to background or inhibit reaction efficiency. Purification can be especially important when working in quantitative applications. For accurate quantification, it is essential to know the exact number of copies that are added to a reaction. Only by purification can you ensure that all oligos added to a reaction are representatives of the target sequence. Absolute Pures offer higher amplification efficiencies, lower background, and better batch-to-batch consistency than oligos that have only been desalted. Chemically pure oligos allow for consistent standard curves and more consistent Ct values over larger numbers of reactions.
Presence of primer dimers and non-specific amplification products can affect quantitative PCR performance in various ways. Non-specific products during dye-based detection assays will contribute to fluorescence levels in the same way as the specific PCR product. This contribution causes a lower threshold cycle value and an overestimation of the initial concentration of the target nucleic acid. During probe-based assays, non-specific products do not contribute to fluorescence unless they also contain the probe sequence. Probe sequences increase specificity of the PCR reaction, but non-specific products will consume resources (such as enzyme, dNTPs, and primers) that could have been used to amplify the target of interest. Probe-specific products will therefore reduce the efficiency of a PCR reaction, potentially leading to false negatives when detecting low concentrations of nucleic acid. Non-specific products create ambiguity in melting curve analyses by introducing extra peaks or increasing the size of peaks present. Primer dimers can outcompete low concentration templates during the reaction because they are present in higher concentrations. This higher concentration can allow primer dimers to be amplified more efficiently than the target of interest leading to false negatives or quantitation that is significantly off.
Non-specific amplification and primer dimers reduce sensitivity. Non-specific products and primer dimers compete for amplification of the specific target which will reduce sensitivity. Primer dimers or primers annealing to non-specific targets with high affinity will form dsDNA products that will have a low melting temperature and contribute to high background fluorescence that can obscure specific target detection. Primer dimers are especially troublesome at low copy numbers because primer dimers will have a higher amplification efficiency compared to the product of interest. At low copy numbers there are fewer targets so there will be more primer annealing to primer rather than target/primer since it is more concentratorion dependent. False positive results can occur in reactions where there is no target present. False positives are more common when using an intercalating dye since it will stain any dsDNA.
Primer dimers and non-specific products can negatively impact quantitative results by altering linearity of the standard curve and efficiency of amplification over the course of its range. Accumulation of these products prior to the first few cycles results in greater than zero fluorescence intensity, which can lead to crossing of the threshold earlier than the target amplification causing inaccuracies in quantitation. This also narrows the window for accurate quantification making it difficult to tell the difference between samples of high and low abundance. Non-specific products that are similar in size to the amplicon can go undetected by melt curve analysis or agarose gel electrophoresis. Either of these factors can lead to inaccurate quantitation of expression ratios or DNA copy number. This is especially important when small differences in efficiencies become magnified during fold-change calculations in relative quantification experiments.
Avoidance of primer dimer and non-specific amplification can be approached on several levels. At the most basic level one can attempt to computationally avoid self-complementarity within each primer as well as complementarity between primers. Designing primers such that the ends of the primers do not have complementarity also helps avoid primer dimer formation. Further, experimental conditions can be optimized to discourage primer dimer formation by decreasing the concentration of primers, thus decreasing the likelihood that primers will find each other by sheer volume (effective molarity), and decreasing Mg2+ concentrations which will slow polymerase activity while increasing stringency of binding. Additionally, performing the PCR reaction at higher temperatures will discourage formation of primer dimers due to more discriminatory annealing.
Primer design is crucial to specificity of the PCR reaction. Primer should be checked to ensure there is no self-complementarity at the 3' end. When setting up PCR primers it is important that the forward and reverse primer are not complementary to each other, even slightly, as they will then have the ability to bind to each other and create a stable double stranded DNA. This will then go through PCR, creating duplicates of that specific DNA segment. Primer should also be checked for self-complementarity that could cause the primer to fold back on itself creating a hair loop. Hair loops should not be extremely stable or unfoldable as that might hide the region of the primer that is supposed to bind. GC content should be checked to not be too high or too low. Same with the melting temperatures of each primer; they should be roughly equal. Checking the specificity will ensure that there are no known repeats or genes that have similar sequences to the primer. Primer should also be checked for long repeats of one nucleotide.
Reaction conditions and stoichiometry can also be optimized to alter the tradeoff between efficiency and specificity. Primer concentrations can be decreased to limit self-dimers and non-specific binding due to the effects of mass action; optimizing the lowest possible concentration that allows detection of the target sequence can decrease primer-dimers while maintaining sensitivity. Optimizing Mg2+ concentration can also improve specificity as higher concentrations can stabilize non-specific binding and increase enzyme activity on non-target sequences. Conversely, too little Mg2+ can decrease specificity. The annealing temperature can also be adjusted to a higher temperature (as high as the Ta can tolerate) to help increase specificity, or a touchdown PCR can be run to help favor specificity in the initial cycles. Additives can also be included to help increase specificity and stability of the desired duplex. Ensuring that the buffer maintains its pH can also help increase enzyme specificity.
Table 3 Comparative Strategies for Preventing Primer Dimers and Non-Specific Amplification
| Strategy Category | Mechanism of Action | Primary Benefit |
| Architectural Design | Elimination of terminal complementarity and hairpin structures | Prevents formation of dimerization substrates |
| Stoichiometric Optimization | Reduction of primer concentration to minimal effective levels | Diminishes mass-action driven inter-primer associations |
| Thermal Stringency | Elevated annealing temperatures or touchdown protocols | Enforces discriminatory binding kinetics favoring specific targets |
| Temporal Enzymatic Control | Hot-start polymerase activation mechanisms | Prevents low-temperature mispriming and extension |
| Chemical Environment | Balanced magnesium and buffer composition | Modulates duplex stability and polymerase fidelity |
Primer dimers and non-specific amplification are often linked to both design limitations and oligonucleotide quality. While thermocycling conditions and reaction optimization play a role, primer sequence characteristics and synthesis purity are frequently the underlying contributors. Our approach to minimizing non-specific amplification includes:
By integrating careful design principles with controlled synthesis and purification strategies, the risk of primer dimer formation and non-specific amplification can be significantly reduced.
Persistent primer dimers, unexpected amplification bands, elevated baseline fluorescence, or inconsistent Ct values may indicate that primer design or quality requires further evaluation. A technical review may be beneficial if you are working with:
Addressing primer sequence parameters together with synthesis and purification considerations can reduce troubleshooting time and improve amplification reliability. Contact our scientific team to discuss your primer design and synthesis requirements and determine the most appropriate strategy for minimizing non-specific amplification in your application.
References
Unintended primer–primer amplification products.
They consume reagents and generate background signal.
Complementary regions promote primer–primer binding.
Yes, it removes truncated and heterogeneous primers.
Through optimized design, concentration, and high-quality synthesis.


 Loading ......
Loading ......