Quantitative PCR's sensitivity relies directly on the quality of the synthetic oligonucleotides used. These oligonucleotides serve as the recognition elements that determine how well rare targets are transformed into quantifiable amplicons. Poor quality primers or probes create variation that is amplified during PCR, resulting in increased background, lower efficiencies, and unstable Ct values. Incomplete synthesis products, modifications, and damaged bases are inhibitors of hybridization and polymerase progression. The increased Ct value lowers the sensitivity of your qPCR reaction. For that reason, high quality oligonucleotide preparations are required for optimal sensitivity when quantifying rare events such as low copy number transcripts, minimal residual disease or low-copy infection.
 Principle of one step RT-qPCR process in primer immobilized network (PIN) microparticles1,5
Principle of one step RT-qPCR process in primer immobilized network (PIN) microparticles1,5
The sensitivity of qPCR reactions is contingent on the quality of the oligonucleotides used. Because oligos dictate target specificity and amplification efficiency, impurities in oligonucleotides can drastically reduce sensitivity. If an oligo is degraded, it can bind to the template without allowing extension of the polymerase. These primers essentially compete with intact oligos, while wasting reagents. Synthesis impurities (remaining coupling reagents, solvents, etc.) can poison the enzymes used for reverse transcription and amplification, which decreases amplification efficiency of template molecules. Modifications that induce sequence heterogeneity into the bases or backbone (oxidation, etc.) can decrease binding affinity by increasing specificity. This reduction in efficiency can force users to use greater quantities of template in order to observe detection events, meaning any rare templates have a lower chance of being observed.
Table 1 Comparative Impact of Oligonucleotide Quality Parameters on qPCR Analytical Performance
| Quality Parameter | Impact on Sensitivity | Consequence of Compromised Quality |
| Chemical Purity | Elevated background from contaminants obscures low-copy detection | False negatives; overestimated detection limits |
| Sequence Integrity | Truncated species reduce effective primer concentration | Loss of linearity; unreliable copy number estimation |
| Structural Stability | Degradation diminishes binding affinity for rare targets | Temporal sensitivity loss; batch-to-batch inconsistency |
| Specificity Architecture | Off-target binding generates competing amplification | False positives; compressed dynamic range |
Limit of Detection (LOD) is the lowest amount of target that can be distinguished from background levels. This aspect is controlled by the purity of the oligonucleotides used. A contaminated oligonucleotide will increase the background fluorescence (primer dimers and non-specific products), hiding the initial cycles where the real product is accumulating. The threshold cycle will be delayed causing a higher Ct to be reported. For low abundance targets this may mean the difference between an incorrect number of copies or a negative result. Accuracy can also be affected if the oligonucleotide used degrades from batch to batch. This changes the affinity of the oligonucleotide to the target sequence causing inconsistent standard curves. This will negatively affect relative quantification. Chemical consistency will allow the accumulation of fluorescence to be directly proportional to the amount of template present.
Stability of the oligos during thermal cycling determines how many oligos are available for hybridization and enzymatic reaction. Degradation of oligos due to depurination, oxidation, and hydrolysis will produce shorter oligos that may still bind to their target but will not be amplified efficiently. These degraded oligos will bind to template molecules, wasting reagents and reducing the signal produced. Degradation of probe reporter groups can also occur (bleaching, chemical degradation), resulting in a lower signal output and decreasing the signal to background. Oligo stability can be maintained by storing them properly and by modifying the oligo with stabilizing chemistry. Stable oligos will have uniform performance over many cycles.
Quality parameters which can be important for diagnostic uses of synthetic oligonucleotides include sequence fidelity, length, purity, and stability. Sequence fidelity can impact hybridization of the oligonucleotide to its intended target sequence without mismatches that could otherwise generate false negatives or contribute to cross-reactivity. Length ensures that the oligonucleotide was fully synthesized rather than stopping part way through synthesis. Purity ensures removal of residual phosphoramidites, organic molecules, and protecting groups leftover from synthesis which may impact enzyme activity. Stability can refer to stability of the phosphodiester linkages as well as stability of any modifications present on the oligonucleotide. All of these parameters impact performance of an oligonucleotide by influencing its ability to hybridize to target sequences, be amplified by polymerases, and produce a detectable signal. For this reason, these characteristics are commonly validated during the manufacturing process.
The accuracy of the sequence should be defined as the oligonucleotide incorporating only those nucleotides that are present in the designed sequence, as confirmed by an orthogonal technique such as mass spectrometry or sequencing. A single base change or swapping of bases incorporated as a result of synthesis inefficiencies can lead to loss of target detection or non-specific binding to off-target sequences. Accuracy of length refers to the amount of full length target sequence versus truncated species within the synthesized population. Incomplete coupling of nucleotides during solid phase synthesis will lead to creation of products missing the terminal nucleotide or nucleotides (N-1, N-2 etc.) and elongated products as a result of over-coupling (N+1). These shorter and longer products will take up binding real estate but will not be efficiently extended by a polymerase resulting in a lower effective concentration of the desired primer or probe. Furthermore this will vary from synthesis to synthesis.
Chemical purity refers to the removal of leftover reagents used in the chemical synthesis of DNA, such as acetonitrile, oxidizing agents, or protecting groups, as well as organic solvents that may have been transferred during the cleavage and deprotection reaction conditions. These materials can interfere with the ability of reverse transcriptase and DNA polymerases to function properly or create high background signals that interfere with specific detection. Consistency means that the chemical characteristics and behaviors of oligonucleotides should be the same from batch to batch. If one synthesis run uses phosphoramidites of differing quality, or couples or purifies to different extents than another synthesis run, the lot-to-lot variability is observed as changes in melting temperature, amplification efficiency and non-reproducible standard curves. Purification by chromatography separates fully intact product from degradation products (shorter oligomers) as well as chemical impurities. Only compounds that are the correct size (mono-disperse) and chemistry are included in the reaction to provide quantitative accuracy and reproducibility between assays.
Incomplete or truncated DNA sequences and synthesis impurities adversely affect qPCR. The phosphoramidite method of DNA synthesis produces incomplete sequences that lack a 3'-end nucleotide as well as the desired complete sequence. In addition, deprotection reagents, amplification inhibitors such as nucleotide protecting groups, and organic solvents remain in the sample if it is not completely purified. These incomplete DNA fragments and impurities will compete with your target oligonucleotide for enzyme interaction and also bind reaction components, creating background noise that makes it difficult to distinguish your desired product. As a result of these issues, your qPCR results may show a lower than expected efficiency, increased Ct value, and inconsistent data.
Incomplete extension products that result from inefficient coupling steps will lead to truncated oligonucleotides that are no longer fully complementary to the template but still bind to the template partially. These truncated primers can bind to template sequences, decreasing overall efficient binding of primers to templates and effectively decreasing polymerase activity since these shorter sequences are unable to be extended efficiently. While these primers are bound to the template, the template cannot bind to a fuller-length primer. In the case of probes, these truncated probes can hybridize to the target sequence without producing any fluorescence signal. The decrease in the number of probes able to produce a signal increases the Ct value. In situations where you are detecting low amounts of nucleic acid, these incomplete extension products can be significantly abundant. This makes it hard to determine if your template will bind to a full-length primer or not since they can be in similar concentrations.
Impurities remaining from synthesis of the oligonucleotide such as dye impurities, incomplete deprotection byproducts, or organic solvent residues contribute directly to background fluorescence. Carryover of fluorescent dye impurities due to incomplete purification adds background signal that cannot be distinguished from probe degradation or product accumulation. Impurities remaining from incomplete deprotection and synthesis may interact nonspecifically with enzymes or other buffer components, contributing to background fluorescence or inhibiting specific interactions necessary for signal production with probes. Background fluorescence narrows the window between negative and positive results, making it more difficult to detect small amounts of target sequence.
The performance of qPCR relies heavily on the purity of the primers involved. Many methods exist for purification of oligonucleotides, which strongly affect the ratio of target to deletion products present in a primer sample. Desalting only removes salts and leftover reagents from the synthesis process. However, failure products (usually deletions) are not removed and will compete with the primer for reaction efficiency. Size exclusion HPLC separates full length strand from deletion products through hydrophobic interactions and can greatly improve purity. PAGE separation goes one step further, separating strands by charge to mass ratio. Purification Method has been shown to greatly affect Ct values and minimum detectable copy number. For applications requiring single copy sensitivity or extreme linearity, a high resolution purification method should be used.
Desalted (dUTP triphosphates) products include all molecules synthesized (including truncated products due to incomplete coupling efficiencies). These truncated products contribute to increased background fluorescence (due to primer dimers and non-specific priming events) making detection of low copy targets difficult. HPLC purified products separate full-length product from truncated fragments due to differences in hydrophobic interactions. This results in a much "cleaner" preparation with less background amplification and improved signal to noise ratio. PAGE purified products are sized separated and represent the most effective way to isolate a single population of full-length product. This can be especially important for longer oligonucleotides which have increased batch to batch variation due to the increased number of couplings reactions involved in their synthesis. Purified oligonucleotides are required for quantitative applications although desalted oligonucleotides can be used for traditional endpoint PCR.
Chemical modifications to probes including the addition of reporter/quencher groups or backbone modifications further complicates purification method choice as some methods may damage probe integrity/function. While chromatography methods can often be tweaked (change in mobile phase conditions) to elute probes with a variety of modifications, very hydrophobic adducts may need special resins to facilitate purification from truncated non-labeled species. Electrophoretic methods can offer much higher resolution, but modifications that impact migration may not be cleanly separated. Additionally, some modifications may not be stable under the denaturing conditions needed for proper size separation. Other purification methods use acidic/basic conditions that could destroy acid-labile chemical modifications or cause depurination in longer sequences. As such method choice can also depend on what chemical modifications have been made to the probe and if they will stand up to the purification method of choice given the needed level of purity.
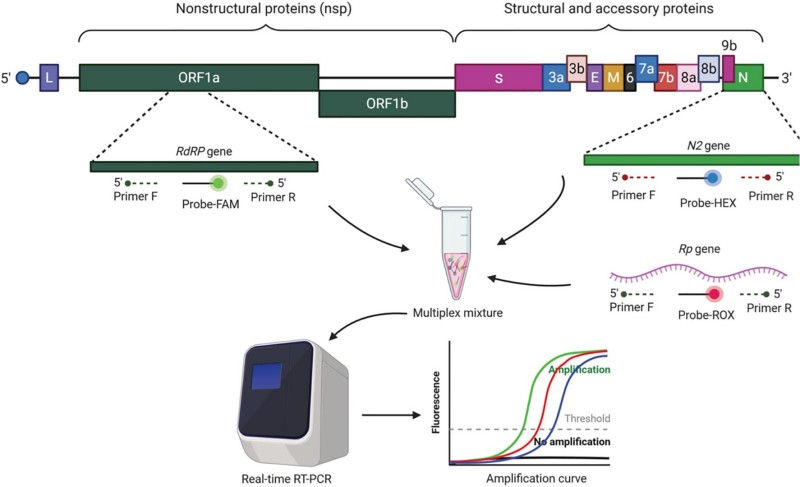 Genome structure of SARS-CoV-2 and the targeted genes in multiplex rRT-PCR assay2,5
Genome structure of SARS-CoV-2 and the targeted genes in multiplex rRT-PCR assay2,5
Batch-to-batch variation is the consistency of oligonucleotide characteristics between manufacturing campaigns. Characteristics include accuracy of sequence and length, chemical purity and functional performance. Batch-to-batch reproducibility is important for quantitative PCR products because variations between lots can result in systematic errors that affect longitudinal studies and comparisons made between laboratories. Variations between synthesis lots may occur because of differences in phosphoramidite coupling efficiencies, purification differences or variations in deprotection conditions. These differences may result in different amounts of full-length product vs. truncated material between batches. As the characteristics of each lot vary, the slope and y-intercept of the calibration curve will change. This will affect the conversion of threshold cycles to copy number for that particular lot.
Batch effects in oligonucleotides directly affect the shape of the standard curve by impacting amplification efficiency and background fluorescence. The slope of the standard curve (expected to be -3.32 with perfect doubling efficiency) varies considerably from lot to lot, especially when observing cartridge purified vs chromatographically purified lots. These differences affect the perceived copy number of samples being tested. Variation in the y-intercept (expected to be zero Ct value at 1 copy) also occurs between lots as a result of differences in primer concentration as well as background fluorescence from remaining synthesis byproducts. Variations in slope and y-intercept can be especially impactful for assays that rely on determining the absolute number of copies because it can cause biased values that exceed laboratory defined thresholds for medical diagnostics or reporting standards.
Table 2 Manifestations of Batch-to-Batch Variability in qPCR Analytical Performance
| Variability Source | Impact on Calibration Parameters | Consequence for Quantification |
| Slope Variation | Altered amplification efficiency between batches | Inconsistent copy number estimations; failed efficiency criteria |
| Y-Intercept Drift | Changed Ct values at reference concentrations | Systematic bias in absolute quantification |
| Purity Differences | Variable background fluorescence and inhibition | Reduced signal-to-noise ratios; delayed threshold crossing |
| Truncation Heterogeneity | Variable effective primer concentration | Irreproducible sensitivity between assay runs |
Synthetic oligonucleotides are characterized by several metrics: sequence identity, purity, concentration, and integrity. Identity ensures that the ordered sequence is the sequence that is received. This is usually guaranteed by analyzing the final product by mass spectrometry or through chromatographic techniques that separate full product from side products generated during synthesis. Purity indicates how much of the oligonucleotide is useable as compared to incomplete sequences (synthesis fail sequences), leftover phosphoramidites, and organic solvents leftover from the cleavage reaction. Concentration is important to know because it affects how much of each reactant you add to your reaction. If the concentration is lower or higher than what is indicated, you are no longer adding the correct ratio of primers to template. Integrity ensures that there have been no modifications, backbone degradation, or other issues that would affect how the oligonucleotide interacts with its target molecule. All of these parameters become critical when performing quantitative PCR since the Ct value is only linearly related to the amount of starting template if the efficiency of the reaction is constant between all runs and oligos used (including batch-to-batch variations and storage times).
Quality control of oligonucleotides can be assessed using several orthogonal methods that confirm different properties of the molecules. Ion-pair reversed-phase HPLC can separate full-length product from degradation products due to differences in hydrophobicity and measure the percent purity using UV absorbance at 260 nm. Mass spectrometry can unambiguously confirm the identity of an oligonucleotide by measuring the accurate mass of the molecule (identifying the parent ion and common charge states) and can also reveal any errors in synthesis or modifications. Size separation using capillary electrophoresis can also be used to separate molecules based on size which can be especially useful for assessing purity of longer oligonucleotides. Typically, full-length product should be the majority species present and the level of impurities from synthesis should be low enough that it will not disrupt enzyme function or cause background issues. If being used for quantitative purposes it should also be confirmed that the oligonucleotide being used is within an acceptable range of amplification efficiency and is specific to the target sequence using melt curve analysis or gel electrophoresis to verify amplicon size.
Deciding which oligonucleotide quality you want to use is also often dictated by the resources available to you and your desired application. You need to ask yourself: What is my intended use? Am I developing something where I have the luxury of trial and error? Will this be for diagnostic use? If so, will it be regulated? Understanding the answers to these questions will help you understand how well your oligo needs to be characterized. Ask yourself how important batch-to-batch consistency is for your experiment. Can you perform your assay multiple times over the course of a year or will you need to run it all within a short period of time? How stringent are your regulatory requirements? If you're just screening or optimizing, you may be okay with using nucleotides that are of lower purity. However, if you are trying to quantitatively detect low abundance targets, you'll want highly purified oligos with quality controls that can reproducibly generate a standard curve.
Explorer grade oligonucleotides are designed for discovery use where confirmation of identity may be the goal along with other objectives such as assay development or qualitative screening. Some variability is permissible for these oligonucleotides in exchange for decreased turnaround time and price. Clinical grade oligonucleotides require high purity via HPLC separation of full length product, extensive analytics, and stringent manufacturing that is consistent batch-to-batch for use in making clinical decisions. Final compliance grades (examples may include assays for IVIV diagnostics or drug products) require fully validated quality systems and regulatory compliant manufacturing and analytics with 100% traceability from beginning materials all the way to release testing. As you increase the grade of oligonucleotide your risk of batch-to-batch variation and premature sequences goes down but at a higher price point and longer turnaround time. Each grade should be carefully considered based on the risk and penalty for failure.
Application dependent information accompanies the product as well. Traceability documents are required based on application needs. Test certificates for research applications may include only verification of identity and approximate concentration. Analytical batch-specific records (control data) can include process parameters, purity information, and stability information for diagnostics applications. Validated methods, forced degradation data, product that has been manufactured with controls in place to ensure the process is robust enough to pass regulatory review may be required for filings. The goal of these documents allows the quality of an oligonucleotide to be reverified, allows analytical performance to be traced to a lot of product, and allow the supply chain of these products to be traced through their lifetime ensuring confidence in any downstream quantitative measurements made.
Oligonucleotides of diagnostic quality are required when test results are used for clinical decision-making, regulatory approval, or directly affect patient safety. Many diagnostic applications require detection of low-frequency alleles or very low copy number targets such as minimal residual disease or rare infections. In these assays there may be little margin for error and decreased primer performance may result in a false negative result. Analytical sensitivity also becomes critical when results are used for diagnostic assays demanding absolute quantification, monitoring over time, or reporting results between laboratories. Validation of production practices such as high-resolution purification methods (e.g., chromatography), sequence verification, and batch consistency become important when diagnostic oligonucleotides are used. Validation to meet diagnostic standards is not necessary if the oligonucleotides are being used for discovery purposes only. However, it is always recommended that any assays developed using discovery oligos be validated with diagnostic grade oligonucleotides before used for clinical testing.
Detection of low-copy transcripts in a clinical setting, or low-genome copies for pathogens requires very high quality oligonucleotides to increase signal-to-background ratio and reduce background amplification. Targets of low abundance may undergo stochastic amplification, which means amplification efficiency of oligos must be as close to 100% as possible to ensure reproducible detection above the cut-off value. Inexperienced trimmed oligonucleotides or buffer contaminants from less pure oligos will bind to targets instead of perfectly matched oligos thus increasing your detection threshold and hiding samples that would otherwise test just above positive. Ensuring you have diagnostic oligos eliminate or greatly reduce the possibility that your reaction components will be used up by forming primer dimers or non-specific priming. Diagnostic oligos are especially important when quantifying virus or rare genomic DNA sequences such as ctDNA.
Diagnostic-grade reagents are often required in regulated laboratories to meet quality system requirements and reduce liability associated with incorrect test results. Documentation such as certificates of analysis, lot release records, and stability documentation provide chain of custody and allow traceability/recalls to be performed should any issues arise. Diagnostic-grade production lots are also easier to submit to regulatory agencies and conduct audits on. Research-grade reagents lack supporting documentation and are therefore ineligible to be used clinically or commercially as a diagnostic assay. If the outcome of an assay would impact patient care by delaying treatment, provide false sense of security, or cause the patient to receive unnecessary treatment based on incorrect drug levels or copy number then diagnostic grade starting materials are considered first.
Table 3 Quality Grade Selection Criteria Based on Application Requirements
| Application Context | Purity Requirement | Documentation Level | Consequences of Suboptimal Quality |
| Exploratory Research | Moderate; cartridge purification acceptable | Basic identity confirmation | Failed optimization experiments; preliminary data requiring validation |
| Clinical Diagnostics | High; chromatographic purification mandatory | Comprehensive batch records and stability data | False negative/positive results; misguided therapeutic decisions |
| Regulated Assays | Very high; stringent impurity profiling | Full regulatory dossier with validated methods | Regulatory rejection; legal liability; patient safety compromise |
| Low-Abundance Detection | Maximum; minimal truncation heterogeneity | Detailed purity profiling and functional validation | Missed rare variants; underestimated pathogen load; delayed diagnosis |
qPCR sensitivity depends not only on primer and probe design, but also on oligonucleotide purity, sequence accuracy, and batch consistency. Truncated products, synthesis-related impurities, or heterogeneous probe populations can compete with functional oligonucleotides and reduce amplification efficiency. Our capabilities in qPCR oligonucleotide synthesis include:
By aligning synthesis precision, purification control, and analytical validation with qPCR performance requirements, detection sensitivity and reproducibility can be significantly improved.
If your qPCR assays show reduced sensitivity, inconsistent Ct values, elevated baseline fluorescence, or variability across batches, oligonucleotide quality may be a contributing factor. A technical evaluation may be particularly valuable when working with:
Optimizing primer and probe quality alongside design parameters can reduce variability and improve detection limits in demanding qPCR applications. Contact our team to discuss your qPCR oligonucleotide requirements and determine the most appropriate synthesis and purification strategy for your assay.
References
Impurities and errors reduce amplification efficiency and signal-to-noise ratio.
They increase background and reduce detection accuracy.
Yes, HPLC- or PAGE-purified oligos improve sensitivity.
Yes, it impacts reproducibility and standard curves.
For low-copy targets and diagnostic qPCR assays.


 Loading ......
Loading ......